
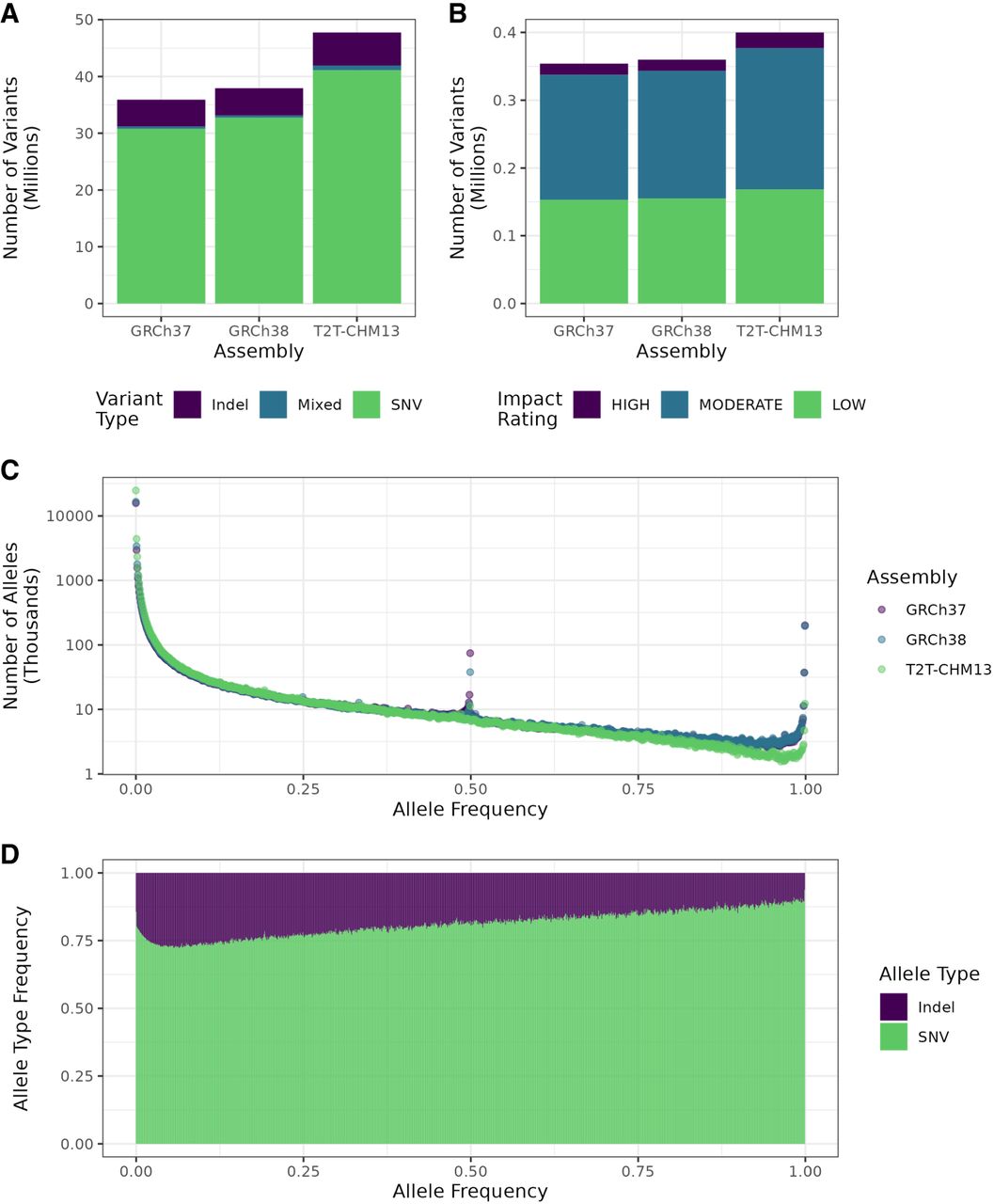
Figure 4.
Summaries of variant characteristics from the whole cohort. (A) Comparison of overall variant counts, separated by variant type (SNV, indel, or mixed, i.e., both) between variant calls from the different assemblies. (B) Overall numbers of variants by predicted impact in the whole call set as determined by SnpEff for each assembly. Variants with impact rating MODIFIER were excluded. (C) Distribution of allele frequencies in the three assemblies. (D) Fraction of each variant type by allele frequency using T2T-CHM13. Frequencies were rounded to the closest multiple of 0.001 (1/1000).








