
Graph-based self-supervised learning for repeat detection in metagenomic assembly
Abstract
Repetitive DNA (repeats) poses significant challenges for accurate and efficient genome assembly and sequence alignment. This is particularly true for metagenomic data, in which genome dynamics such as horizontal gene transfer, gene duplication, and gene loss/gain complicate accurate genome assembly from metagenomic communities. Detecting repeats is a crucial first step in overcoming these challenges. To address this issue, we propose GraSSRep, a novel approach that leverages the assembly graph's structure through graph neural networks (GNNs) within a self-supervised learning framework to classify DNA sequences into repetitive and nonrepetitive categories. Specifically, we frame this problem as a node classification task within a metagenomic assembly graph. In a self-supervised fashion, we rely on a high-precision (but low-recall) heuristic to generate pseudolabels for a small proportion of the nodes. We then use those pseudolabels to train a GNN embedding and a random forest classifier to propagate the labels to the remaining nodes. In this way, GraSSRep combines sequencing features with predefined and learned graph features to achieve state-of-the-art performance in repeat detection. We evaluate our method using simulated and synthetic metagenomic data sets. The results on the simulated data highlight GraSSRep's robustness to repeat attributes, demonstrating its effectiveness in handling the complexity of repeated sequences. Additionally, experiments with synthetic metagenomic data sets reveal that incorporating the graph structure and the GNN enhances the detection performance. Finally, in comparative analyses, GraSSRep outperforms existing repeat detection tools with respect to precision and recall.
Metagenomics is a scientific discipline that involves analyzing genetic material obtained from complex uncultured samples housing DNA from diverse organisms (Wooley et al. 2010). This field utilizes high-throughput sequencing and bioinformatic techniques to characterize and compare the genomic diversity and functional potential of entire microbial communities without the need for isolating and culturing individual organisms (Yang et al. 2021). The resulting data can provide insights into the ecological roles and evolutionary relationships of the microorganisms present in the sample (Schatz et al. 2010).
However, the sequencing of DNA from such samples poses unique challenges. One of the major challenges in the metagenomic assembly is the presence of repeats (Ghurye et al. 2016; Lapidus and Korobeynikov 2021), which are sequences of DNA that are similar or identical to sequences elsewhere in the genome (Treangen and Salzberg 2012). The challenges posed by repeats in isolated genomes have primarily been addressed through the use of long-read technologies (Koren and Phillippy 2015). However, metagenomics presents a more complex problem as microbial mixtures often contain multiple closely related genomes that differ in just a few locations owing to structural variants (Martin et al. 2023), such as horizontal gene transfer (Soucy et al. 2015), gene duplication, and gene loss/gain (Iranzo et al. 2019). Reads spanning the length of individual strains are required to fully resolve these genome-scale repeats present in microbiomes.
These repetitive elements, although natural and abundant in genomes, complicate the process of genome assembly and comparison (Treangen et al. 2009). They intricately tangle the assembly graph, making it difficult to distinguish the order, orientation, and copy number variation of genomes comprising the microbiome under study, resulting in fragmented assemblies. Moreover, repeats introduce ambiguities for comparative genomics, hindering differentiation between identical or similar regions and complicating the understanding of gene functions, regulatory elements, and their role in genetic disorders (Treangen and Salzberg 2012). To overcome these obstacles, the precise identification and annotation of repeated sequences is necessary. Unraveling the complexities of repeated sequences is not only crucial for enhancing genome assembly but also essential for deciphering intricate regulatory mechanisms and evolutionary processes. Indeed, identifying these repeats is foundational for understanding genome stability, gene expression, and disease susceptibility, making the development of accurate repeat detection methods vital for advancing genomic research (Girgis 2015).
Graphs are powerful tools for visualizing complex relationships between various objects, such as DNA sequences. Graph-based algorithms can effectively represent the interconnections and overlapping patterns within genomes (Koutrouli et al. 2020), in which the nodes in the graph represent unique DNA sequences. Because of the tangled nature of repeated sequences within the assembly graph, exploiting graph structure becomes particularly advantageous. As an illustrative example, Figure 1 portrays the assembly graph obtained from a simulated metagenome with two organisms. In this scenario, three random sequences are generated. Two of these sequences are inserted as intra-genome repeats in each organism, whereas the third one is inserted in both organisms, serving as an inter-genome repeat. This graph is visualized using Bandage (Wick et al. 2015), in which the length of each node is proportional to the length of the corresponding contig. A node labeled as a repeat (which is colored red in the figure) represents a unique DNA sequence that occurs in several positions of the metagenome sample. The graph reveals that repeats are represented by central and well-connected nodes, indicating the potential of utilizing the inherent graph structure in genomic data for identifying repeated sequences. However, graph structure is usually not enough to tell apart the repeat nodes from some of the nonrepeat ones. This motivates an approach that combines graph features with sequencing information such as read coverage or length of the DNA sequence.
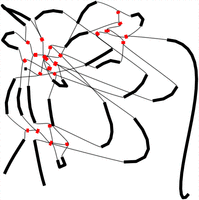
Assembly graph representation with repeat contigs in red.
Previous studies have employed prespecified graph features in combination with machine learning techniques to address the challenge of detecting repeats, treating it as a node classification problem (Ghurye and Pop 2016; Ghurye et al. 2019). In this context, the nodes of the graph represent DNA sequences, and the objective is to classify them into repeats and nonrepeats. However, given the vast amount of genomic data, there remains ample opportunity for enhancement through learning discriminative graph features. One of the promising ways to achieve this is by employing graph neural networks (GNNs) (Wu et al. 2020). GNNs have the unique ability to learn distinctive and valuable features for the nodes within the graphs. Unlike predefined features, GNNs generate these characteristics through trainable iterative computations, making them adaptive to the specific data. These features have shown promising results in many other fields (Chowdhury et al. 2021; Čutura et al. 2021; Glaze et al. 2023; Zhao et al. 2023), emphasizing the efficiency of utilizing GNNs to classify nodes accurately and uncover the complexities within large graphs (Hamilton et al. 2017).
However, one of the primary challenges in genomic data analysis is the fact that most of the data are unlabeled, particularly in distinguishing between repeat and nonrepeat sequences. This characteristic of the data prevents the application of supervised or semisupervised learning techniques for classifying DNA sequences (Kipf and Welling 2017). In the absence of labeled data points offering insights into each class, these conventional methods become ineffective. To overcome this issue, self-supervised learning emerges as a natural and powerful alternative to leverage the vast unsupervised data (Jaiswal et al. 2020). In self-supervised learning, specific data points (nodes) are initially given (potentially noisy) labels. Subsequently, machine learning algorithms are employed, coupled with fine-tuning steps, to refine the model's performance. This approach ensures the ability to classify data points without requiring access to their true labels.
In this paper, we propose GraSSRep, a novel graph-based algorithm to identify and detect the repeated sequences in the metagenomic assembly in a self-supervised manner. Our contributions are threefold: (1) By leveraging GNNs, we devise the first method that learns (rather than prespecifies) graph features for repeat detection; (2) we establish the first algorithm that uses self-supervised learning for repeat detection, leveraging existing methods to generate noisy labels that we then refine and expand using our learnable architecture; and (3) through numerical experiments, we demonstrate the robustness of our methodology, the value of each of its steps, and the performance gain compared with the state of the art.
Methods
Given paired-end reads, our goal is to identify repeated DNA sequences in the metagenome (for the precise criteria used to define a repeat, see the Results subsection “Experimental setup”). An overview of our method specifically designed for this task is illustrated in Figure 2. In the subsequent sections, we provide a detailed explanation of each step involved in the pipeline.
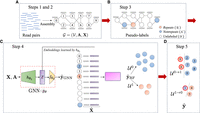
Overview of GraSSRep. (A) Reads are assembled into contigs, forming the nodes of the assembly graph. The graph structure (edges) is provided by metaSPAdes. Also, feature vectors are computed for each contig. (B) Contigs with distinctive sequencing features are selected as training nodes and labeled. (C) The assembly graph is input into a GNN. Embeddings are generated for each contig and combined with the initial features. A random forest classifier predicts labels for all contigs based on the augmented feature vectors. (D) Sequencing features are employed to identify outliers within each predicted class, leading to the reassignment of their class labels.
Step 1: assembly graph construction
In the initial step, we construct an assembly graph in order to leverage graph features for repeat detection. To do so, we
assemble the input reads to contigs and obtain the assembly graph as illustrated in Figure 2A. Here, we use the popular metagenomic assembler, metaSPAdes (Nurk et al. 2017), which employs a multisized de Bruijn graph to derive the contigs and the connections between them. We consider these assembled
contigs as the nodes V of our assembly graph, where 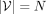 . We denote by
. We denote by 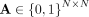 the adjacency matrix of the corresponding unweighted graph, where Aij = 1 if there is an edge between contig i and j, and
the adjacency matrix of the corresponding unweighted graph, where Aij = 1 if there is an edge between contig i and j, and 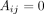 otherwise. Note that assembly graphs can also be generated similarly using alternative metagenomic assemblers like MEGAHIT
(Li et al. 2015) and metaFlye (Kolmogorov et al. 2020), either by assembling the contigs and connecting them using read-mapping information or by directly utilizing the assembly
graph provided by the assembler. We plan to support these additional assembly graph formats in the future. We specifically
choose metaSPAdes because it is a well-known state-of-the-art short-read assembler that is easy to use and offers high accuracy.
The most significant benefit of metaSPAdes is that it provides the assembly graph directly alongside the assembled contigs.
This feature fits seamlessly into our pipeline and allows us to bypass the additional step of read-mapping to identify connections
between contigs. This integrated process not only simplifies our workflow but also accelerates graph construction, making
metaSPAdes the optimal choice for our framework. However, we can alternatively construct our assembly graph manually by assembling
the reads and utilizing read-mapping data; this is discussed in further detail in the section “Alternative graph construction”
in the Supplemental Material.
otherwise. Note that assembly graphs can also be generated similarly using alternative metagenomic assemblers like MEGAHIT
(Li et al. 2015) and metaFlye (Kolmogorov et al. 2020), either by assembling the contigs and connecting them using read-mapping information or by directly utilizing the assembly
graph provided by the assembler. We plan to support these additional assembly graph formats in the future. We specifically
choose metaSPAdes because it is a well-known state-of-the-art short-read assembler that is easy to use and offers high accuracy.
The most significant benefit of metaSPAdes is that it provides the assembly graph directly alongside the assembled contigs.
This feature fits seamlessly into our pipeline and allows us to bypass the additional step of read-mapping to identify connections
between contigs. This integrated process not only simplifies our workflow but also accelerates graph construction, making
metaSPAdes the optimal choice for our framework. However, we can alternatively construct our assembly graph manually by assembling
the reads and utilizing read-mapping data; this is discussed in further detail in the section “Alternative graph construction”
in the Supplemental Material.
Step 2: feature extraction
We compute features of the contigs that are informative in determining which contigs are repetitive. We consider two types
of features: sequencing and graph based. Sequencing features (contig length and mean coverage) are obtained during the sequencing
process before constructing the contig graph and are used in Steps 3 and 5. In addition, we incorporate four graph-based features
that are widely used in the literature: betweenness centrality, k-core value, degree, and clustering coefficient. Previous
studies have emphasized the significance of betweenness centrality (Segarra and Ribeiro 2015) in identifying repeats (Ghurye and Pop 2016). Additionally, KOMB (Balaji et al. 2022) has underscored the crucial role of the k-core value in anomaly detection within contigs. Furthermore, the degree of nodes
indicates their connectivity strength with other contigs, aiding in the identification of repeated regions. We also consider
the clustering coefficient owing to its substantial impact on node classification tasks, as well as its demonstrated positive
effects and favorable outcomes in various related domains (Zaki et al. 2013). We store the graph-based features in a matrix 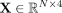 , where every row contains the four graph-based features of a given node (contig) in the graph. Thus, we define our featured
graph of interest as
, where every row contains the four graph-based features of a given node (contig) in the graph. Thus, we define our featured
graph of interest as 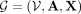 , as shown in Figure 2A.
, as shown in Figure 2A.
Step 3: selection of the training nodes
Recall that we do not have any prior information (labels) on whether any contig is a repeat or not. In this context, the idea of self-supervised learning is first to do a high-confidence classification of a subset of the contigs (assigning potentially noisy labels, denominated pseudolabels, to a subset of the nodes) and then use those nodes as a training set for a machine learning model that can classify the remaining contigs. We generate this set of pseudolabels using the sequencing features from Step 2. In generating pseudolabels, it is important only to consider those for which we have a high level of confidence so that the training process based on these pseudolabels is reliable.
In defining our pseudolabels, we rely on the fact that shorter contigs with higher coverage are highly likely to be repetitive,
whereas very long contigs with lower coverage are more likely to be nonrepeat contigs (Ghurye and Pop 2016). More precisely, let us define as 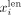 and
and 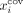 as the length (number of base pairs) and coverage (mean number of reads mapped to the base pairs in the contig) of node i, respectively. We set a percentile p (with 0 ≤ p ≤ 50), based on which we define the following thresholds:
as the length (number of base pairs) and coverage (mean number of reads mapped to the base pairs in the contig) of node i, respectively. We set a percentile p (with 0 ≤ p ≤ 50), based on which we define the following thresholds: 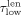 is the pth percentile of the lengths among all contigs in V,
is the pth percentile of the lengths among all contigs in V, 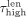 is the (100 − p)th percentile of the lengths among all contigs, and
is the (100 − p)th percentile of the lengths among all contigs, and 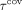 is the (100 − p)th percentile of the coverages among all contigs. Based on these thresholds, we divide the contigs into three sets—the repeats
R, the nonrepeats N, and the unlabeled U—as follows:
is the (100 − p)th percentile of the coverages among all contigs. Based on these thresholds, we divide the contigs into three sets—the repeats
R, the nonrepeats N, and the unlabeled U—as follows: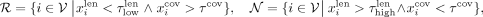 (1)
and
(1)
and 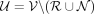 . In Equation 1, contigs shorter than the lower length threshold and with a coverage surpassing the coverage threshold are included in the
training set with a repeat pseudolabel (R). Conversely, contigs exceeding the higher length threshold and having a coverage
below the coverage threshold are added to the training set with a nonrepeat pseudolabel (N). If a contig does not meet any
of these conditions, it suggests that sequencing features alone are not sufficient to determine its classification. Consequently,
these contigs are not included in the training set (U). A simple example of how the assembly graph is divided into three subsets
after this step is depicted in Figure 2B.
. In Equation 1, contigs shorter than the lower length threshold and with a coverage surpassing the coverage threshold are included in the
training set with a repeat pseudolabel (R). Conversely, contigs exceeding the higher length threshold and having a coverage
below the coverage threshold are added to the training set with a nonrepeat pseudolabel (N). If a contig does not meet any
of these conditions, it suggests that sequencing features alone are not sufficient to determine its classification. Consequently,
these contigs are not included in the training set (U). A simple example of how the assembly graph is divided into three subsets
after this step is depicted in Figure 2B.
Step 4: contig classification via self-supervised learning
We leverage self-supervised learning by training a graph-based model on R (binary label of one) and N (binary label of zero) and use that model to classify the nodes in U.
Consider the graph 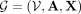 generated in Steps 1 and 2 and denote by
generated in Steps 1 and 2 and denote by 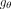 a GNN parameterized by θ (Wu et al. 2020). This GNN takes the graph structure A and the node features X as input and produces labels
a GNN parameterized by θ (Wu et al. 2020). This GNN takes the graph structure A and the node features X as input and produces labels 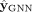 for the nodes at the output. To generate these labels,
for the nodes at the output. To generate these labels, 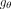 can be viewed as an end-to-end network that is structured as follows:
can be viewed as an end-to-end network that is structured as follows: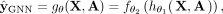 (2)
where
(2)
where 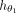 consists of graph convolutional layers followed by an activation function (Agarap 2018). Each layer in
consists of graph convolutional layers followed by an activation function (Agarap 2018). Each layer in 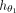 generates new observations for every node based on its neighboring nodes. These convolutional layers are succeeded by
generates new observations for every node based on its neighboring nodes. These convolutional layers are succeeded by 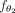 , which represents a fully connected neural network (Haykin 1998). The purpose of this network is to predict the final label for each node based on the features derived from the last layer
of
, which represents a fully connected neural network (Haykin 1998). The purpose of this network is to predict the final label for each node based on the features derived from the last layer
of 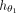 . Note that we provide here a generic functional description of our methodology, whereas in “Experimental setup,” we detail
the specific architecture used in the experiments.
. Note that we provide here a generic functional description of our methodology, whereas in “Experimental setup,” we detail
the specific architecture used in the experiments.
We denote the output of the convolutional layers by 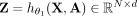 , where d is a prespecified embedding dimension. The ith row
, where d is a prespecified embedding dimension. The ith row 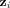 of Z represents new features for contig i, learned in such a way that the final linear layer,
of Z represents new features for contig i, learned in such a way that the final linear layer, 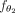 , can predict the class of the contigs based on these features. These embeddings enable us to achieve our objective of understanding
the graph-based characteristics of repeat and nonrepeat contigs. Notice that the features in
, can predict the class of the contigs based on these features. These embeddings enable us to achieve our objective of understanding
the graph-based characteristics of repeat and nonrepeat contigs. Notice that the features in 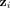 not only depend on graph features of node i but also depend on the features of its local neighborhood through the aggregation of the trainable convolutional layers in
not only depend on graph features of node i but also depend on the features of its local neighborhood through the aggregation of the trainable convolutional layers in
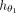 .
.
To learn the parameters 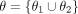 , the GNN undergoes an end-to-end training based on the pseudolabels R and N identified in Step 3. This training process involves
minimizing a loss function that compares the predicted labels
, the GNN undergoes an end-to-end training based on the pseudolabels R and N identified in Step 3. This training process involves
minimizing a loss function that compares the predicted labels 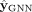 with the pseudolabels
with the pseudolabels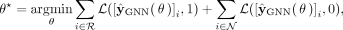 (3)
where L represents a classification loss (such as cross-entropy loss) (De Boer et al. 2005), and we have made explicit the dependence of
(3)
where L represents a classification loss (such as cross-entropy loss) (De Boer et al. 2005), and we have made explicit the dependence of 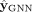 with θ. In essence, in Step 3 we look for the GNN parameters
with θ. In essence, in Step 3 we look for the GNN parameters 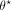 such that the predicted labels for the nodes in R are closest to one, whereas the predicted labels for the nodes in N are closest to zero. Intuitively, the intermediate embeddings Z obtained
using the optimal parameters
such that the predicted labels for the nodes in R are closest to one, whereas the predicted labels for the nodes in N are closest to zero. Intuitively, the intermediate embeddings Z obtained
using the optimal parameters 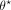 encode learning-based features relevant for the classification beyond the predefined ones in Step 2. Thus, we construct the
augmented feature matrix
encode learning-based features relevant for the classification beyond the predefined ones in Step 2. Thus, we construct the
augmented feature matrix 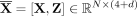 by concatenating the initial graph-based features with those generated by the GNN.
by concatenating the initial graph-based features with those generated by the GNN.
A random forest (RF) classifier is then trained on the pseudolabels 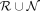 having the augmented features
having the augmented features 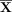 as input. The RF is trained by creating multiple decision trees from different subsets of the data set (a process known as
bootstrapping), with each tree using a random subset of features. When making predictions, the individual trees’ outputs are
combined through majority voting, producing a reliable and precise ensemble model (Breiman 2001). The RF classifier combines the explanatory power of the original graph-based features X found to be relevant in previous
works with the learning-based features Z to generate the predicted labels
as input. The RF is trained by creating multiple decision trees from different subsets of the data set (a process known as
bootstrapping), with each tree using a random subset of features. When making predictions, the individual trees’ outputs are
combined through majority voting, producing a reliable and precise ensemble model (Breiman 2001). The RF classifier combines the explanatory power of the original graph-based features X found to be relevant in previous
works with the learning-based features Z to generate the predicted labels 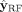 . An overview of how the labels of the training contigs is propagated to all contigs in Step 4 is shown in Figure 2C.
. An overview of how the labels of the training contigs is propagated to all contigs in Step 4 is shown in Figure 2C.
Notice that the sequencing features xlen and 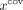 are not used in computing
are not used in computing 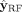 other than in the generation of the pseudolabels. If we were to include these features as inputs to the RF, then the classifier
can simply learn the conditions in Step 1 and obtain zero training error by ignoring all the graph features. This would directly
defeat the purpose of our self-supervised framework. Instead, the current pipeline can distill the graph-based attributes
associated with repeats and nonrepeats, enabling us to generalize this knowledge to classify other contigs effectively.
other than in the generation of the pseudolabels. If we were to include these features as inputs to the RF, then the classifier
can simply learn the conditions in Step 1 and obtain zero training error by ignoring all the graph features. This would directly
defeat the purpose of our self-supervised framework. Instead, the current pipeline can distill the graph-based attributes
associated with repeats and nonrepeats, enabling us to generalize this knowledge to classify other contigs effectively.
Step 5: fine-tuning the labels
In the final step of our method, we enhance the performance of our predictions through a fine-tuning process. We first assign the pseudolabels of the training nodes in R and N as their final predicted labels. Our primary focus is then directed toward the nontraining contigs in U. These contigs have been classified by the RF in Step 4 relying solely on their graph-based features and embeddings learned by the GNN. At this point, reconsidering sequencing features becomes crucial, as they hold valuable information that can significantly contribute to determining the accurate labels of the contigs.
To do so, we divide the contigs in U into two disjoint sets: those predicted as repeats (label 1) by 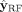 form the set
form the set 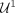 and those predicted as nonrepeats (label 0) by
and those predicted as nonrepeats (label 0) by 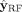 form the set
form the set 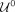 . Within each set, our objective is to identify outliers using the sequencing features xlen and
. Within each set, our objective is to identify outliers using the sequencing features xlen and 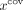 and modify their labels accordingly, similar to Step 3. Within each set, specific thresholds are computed based on the distribution
of sequencing features of the contigs in that set. More precisely, for
and modify their labels accordingly, similar to Step 3. Within each set, specific thresholds are computed based on the distribution
of sequencing features of the contigs in that set. More precisely, for 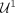 we define
we define 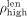 as the (100 − p)th percentile of the contigs’ lengths and
as the (100 − p)th percentile of the contigs’ lengths and 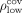 and the pth percentile of the coverage. Conversely, for
and the pth percentile of the coverage. Conversely, for 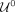 we define
we define 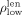 as the pth percentile of the contigs’ lengths and
as the pth percentile of the contigs’ lengths and 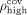 and the (100 − p)th percentile of the coverage. Based on these thresholds, we identify outliers based on the following criteria:
and the (100 − p)th percentile of the coverage. Based on these thresholds, we identify outliers based on the following criteria: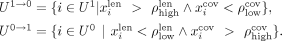 (4)
In Equation 4, we change the label from repeat to nonrepeat (
(4)
In Equation 4, we change the label from repeat to nonrepeat (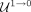 ) for those contigs that are longer than a threshold and have low coverage. Similarly, we change the label from nonrepeat
to repeat (
) for those contigs that are longer than a threshold and have low coverage. Similarly, we change the label from nonrepeat
to repeat (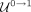 ) for short contigs with high coverage. This process is illustrated in Figure 2D. Notice that we used the same percentile p to compute the thresholds ρ here as that one used to compute the thresholds τ in Step 3. Naturally, we could select a different
percentile here, but we use the same one as this shows good empirical results and reduces the number of hyperparameters.
) for short contigs with high coverage. This process is illustrated in Figure 2D. Notice that we used the same percentile p to compute the thresholds ρ here as that one used to compute the thresholds τ in Step 3. Naturally, we could select a different
percentile here, but we use the same one as this shows good empirical results and reduces the number of hyperparameters.
Summarizing, the final labels 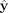 predicted by our model are given by
predicted by our model are given by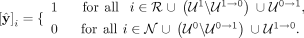 (5)
In Equation 5, we see that the contigs deemed as repeats (
(5)
In Equation 5, we see that the contigs deemed as repeats (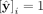 ) by our method are those (1) assigned a repeat pseudolabel in Step 3 (R), (2) classified as repeats by our RF in Step 4 and
not deemed as outliers in Step 5 (
) by our method are those (1) assigned a repeat pseudolabel in Step 3 (R), (2) classified as repeats by our RF in Step 4 and
not deemed as outliers in Step 5 (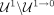 ), or (3) classified as nonrepeats in Step 4 but later deemed as outliers in Step 5 (
), or (3) classified as nonrepeats in Step 4 but later deemed as outliers in Step 5 (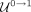 ). Conversely, contigs classified as nonrepeats are those (1) assigned a nonrepeat pseudolabel in Step 3 (N), (2) classified
as nonrepeats by our RF in Step 4 and not deemed as outliers in Step 5 (
). Conversely, contigs classified as nonrepeats are those (1) assigned a nonrepeat pseudolabel in Step 3 (N), (2) classified
as nonrepeats by our RF in Step 4 and not deemed as outliers in Step 5 (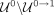 ), or (3) classified as repeats in Step 4 but later deemed as outliers in Step 5 (
), or (3) classified as repeats in Step 4 but later deemed as outliers in Step 5 (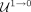 ).
).
Results
In the following sections, we present a comprehensive analysis of our algorithm's performance across various settings.
Experimental setup
Data sets
We test GraSSRep in three types of data sets.
-
Simulated data. To represent distinct organisms, we generate two random backbone genomes with an equal probability of observing each base. Subsequently, a random sequence of length L is generated for each backbone and integrated into the genome with a copy number of C, serving as an intra-genome repeat. Additionally, an inter-genome repeat of length L is randomly generated and inserted C times in both genomes, representing an inter-genome repeat. Unlike the backbone genomes, repeats exhibit a nonuniform distribution of bases, resulting in distinctive characteristics unique to each repeat, setting them apart from the backbone genome. Consequently, we have two genomes, both containing a repeat content of 2 × L × C within a fixed length of 5 million bp for each organism. As a result, the characteristics of the repeats within the genomes can be controlled by adjusting the values of L and C. Finally, simulated reads, each 101 bp in length, are generated using wgsim (https://github.com/lh3/wgsim) with default values for error (2%) and mutation (0.1%).
-
Shakya 1. In this data set, we analyze the reference genomes of a synthetic metagenome called Shakya, which consists of 64 organisms, including 48 bacteria and 16 archaea (Shakya et al. 2013). Based on these reference genomes, read pairs are generated using wgsim, akin to the previous data set. However, unlike the simulated data, all the backbone genomes in this data set are real organisms, containing intricate repeat patterns that are beyond our control. The generated reads are 101 bp long with a high coverage ( ≃ 50) and are produced without any errors or mutations, in order to identify exact repeats in the data.
-
Shakya 2. Read pairs from the Shakya (Shakya et al. 2013) study were obtained from the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number SRR606249, all with a length of 101. We have no influence over coverage or read errors in this set of reads, mirroring real-world settings. This characteristic enables us to evaluate GraSSRep under realistic scenarios.
Assembly
In all experiments, contigs are assembled using the default values of metaSPAdes v3.13.0 for k-mer size, which are k = 21, k = 33, and k = 55. Also, in the error-free case (Shakya 1 data set), we utilize the ‐‐only-assembler option of metaSPAdes and disable the read error correction step.
To assess our model accurately, it is crucial to have the ground-truth labels for the contigs. To identify these labels, all contigs are aligned to the reference genomes using NUCmer (with the ‐‐maxmatch option) (Marçais et al. 2018). Contigs are marked as repeats if they meet specific criteria. Generally, this criterion includes aligning at more than one location with at least 95% identity and 95% alignment length, indicating nonidentical repeats. However, in error-free cases like the Shakya 1 data set, the criterion is aligning at more than one location with 100% identity and 100% alignment length, which indicates exact repeats through the reference genomes.
Method design and hyperparameter choices
To select and label the training nodes, a threshold value p ranging between 30 and 40 is employed in Step 3, depending on the presence of noise in the data. Specifically, p = 35 in instances in which noise is present (simulated data and Shakya 2), ensuring robustness in the presence of data irregularities. However, for noiseless cases (Shakya 1), we set p = 20, leading to a stricter definition of repeat pseudolabels. Previous studies have demonstrated that this choice yields effective repeat detection (Ghurye et al. 2019). However, in the simulated data set, during the fine-tuning step, we observed that setting p = 0 (indicating no need for fine-tuning) yielded superior results. This phenomenon primarily arises owing to the presence of only two organisms in the data set, leading to smaller and simpler assembly graphs. Consequently, the fine-tuning step becomes unnecessary as the labels generated by RF suffice for accurate classification.
In Step 4, the first component of the GNN, 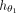 , consists of two consecutive GraphSAGE convolutional layers, each followed by a ReLU activation function (Hamilton et al. 2017). The node representation update in these layers can be mathematically defined as follows:
, consists of two consecutive GraphSAGE convolutional layers, each followed by a ReLU activation function (Hamilton et al. 2017). The node representation update in these layers can be mathematically defined as follows: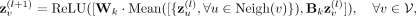 where
where 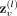 represents the node embedding of the node v at layer l, Neigh(v) represents the set of neighboring nodes of node v, and Mean is an aggregation function that combines the embeddings of neighboring nodes. Moreover,
represents the node embedding of the node v at layer l, Neigh(v) represents the set of neighboring nodes of node v, and Mean is an aggregation function that combines the embeddings of neighboring nodes. Moreover, 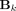 and
and 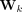 represent the linear transformation matrix for the self and neighbor embeddings, respectively. In this equation,
represent the linear transformation matrix for the self and neighbor embeddings, respectively. In this equation, 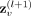 represents the updated embedding of the node v at the next layer (l + 1). Both the first and second convolutional layers have 16 hidden channels. More details on tuning the GNN structure hyperparameters
are provided in the section “GNN hyperparameter tuning” in the Supplemental Material. This results in d = 16 new features being generated for each node, represented as
represents the updated embedding of the node v at the next layer (l + 1). Both the first and second convolutional layers have 16 hidden channels. More details on tuning the GNN structure hyperparameters
are provided in the section “GNN hyperparameter tuning” in the Supplemental Material. This results in d = 16 new features being generated for each node, represented as 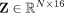 . Because
. Because 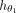 has two graph convolutional layers, the final embeddings combine the features within the two-hop neighborhoods of each node.
Additionally, the second component of the GNN,
has two graph convolutional layers, the final embeddings combine the features within the two-hop neighborhoods of each node.
Additionally, the second component of the GNN, 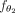 , comprises a single fully connected layer that transforms the newly learned features, Z, into binary classes using a linear
transformation matrix
, comprises a single fully connected layer that transforms the newly learned features, Z, into binary classes using a linear
transformation matrix 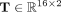 . The GNN is trained for 2000 epochs, utilizing cross-entropy as the loss function and employing the Adam optimizer (Kingma and Ba 2014) with a learning rate of 0.001.
. The GNN is trained for 2000 epochs, utilizing cross-entropy as the loss function and employing the Adam optimizer (Kingma and Ba 2014) with a learning rate of 0.001.
The RF classifier utilizes 100 trees in the forest to generate its results. The split criterion for each decision tree is determined using the Gini impurity measure, ensuring the creation of optimal splits at each node. Finally, to account for the randomness inherent in the training process, both the training and testing steps are repeated for 10 iterations in each case. The reported results are averaged across these iterations, providing a robust and reliable evaluation. As figures of merit, we report the classification accuracy, precision, recall, and F1-score (harmonic mean of precision and recall).
Evaluation on varying repeat characteristics
We leverage the simulated data set introduced in the “Experimental setup” section to examine the effect of three crucial characteristics that are beyond our control within the real data sets:
-
Length of the repeats. To measure the impact of repeat length, we fix the copy number of both inserted intra-genome and inter-genome repeats at C = 25 and vary their length from L = 150 to L = 1000 base pairs, leading to a copy content ranging from 0.15% to 1% in the reference genomes.
-
Copy number of the repeats. We set the length of the inserted repeats to L = 400 base pairs and adjust their copy number from C = 10 to C = 150, increasing the complexity of the data set. This results in a copy content ranging between 0.16% and 2.4% in the reference genomes.
-
Coverage. We generate backbone data by inserting repeats of L = 400 base pairs in length with a copy number of C = 25 to have 0.4% copy content in the reference genomes. The number of generated read pairs is varied, ranging from 0.25 to 2.5 million bp. Consequently, the coverage ranges from five to 50, allowing us to analyze the algorithm's performance under different coverage levels.
These adjustments enable a detailed evaluation of our algorithm's robustness and adaptability across a spectrum of repeat characteristics and coverage scenarios. Note that because of errors and mutations in the generated reads, our analysis considers a repeat as having at least 95% identity over 95% of the length. Consequently, more than just three contigs are identified as repeats in this context, each with copy numbers that may differ from the exact number of inserted repeats.
Because the backbone and inserted repeats are generated randomly in the simulated data sets, we conduct 10 trials for each case to ensure robust results for each condition. Specifically, for each scenario, we generate 10 data sets with the same desired characteristics for repeat length, copy number, or coverage. We then calculate the results for each trial and report the average across these trials for all metrics. Additionally, Figure 3 depicts the error for the F1-score across these 10 trials as a shaded purple area. We use the interquartile range to quantify the error; that is, the error lower bound corresponds to the 25th percentile, and the upper bound corresponds to the 75th percentile across the 10 samples.
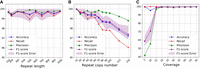
Assessing the method across various repeat characteristics. (A) The model remains stable in metrics even with increasing repeat length. (B) The method is robust to the copy number variation, consistently achieving an F1-score >90%. (C) Higher sequencing coverage improves the model's performance.
As illustrated in Figure 3A, our approach demonstrates resilience to variations in repeat length, with all metrics remaining stable as the repeat length increases. Consistently achieving an average F1-score >99% indicates that our approach can effectively detect repeated contigs even when longer repeats are present in the data set.
Figure 3B shows the performance attained when varying the copy number. Our method consistently achieves an average F1-score exceeding 97%, and for copy numbers below 70, it consistently surpasses 99%. Additionally, the average precision is >99% in almost all cases. However, we observe a decreasing trend in the average recall, which results in a corresponding decrease in the F1-score as the copy number increases. This drop occurs because a higher copy number for the repeats creates a more tangled assembly graph with a lot of connections between the assembled contigs, making it more challenging to detect all the repeats. Note that for copy numbers less than 10, the assembly graph remains untangled, and repeats are detected with 100% accuracy using coverage or degree without requiring additional complex steps.
As demonstrated in Figure 3C, the model's performance exhibits a constant improvement with increased coverage, as expected. Specifically, when coverage is higher than 20 (corresponding to 1 million reads), the model achieves an almost perfect rate of ∼100% for all metrics.
Ablation study of the steps of the algorithm
We focus on the behavior of our method (see Methods) across different steps using the Shakya 1 data set. After assembling
and constructing the graph, we have 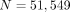 contigs as the nodes of the graph, out of which 13,842 contigs are exact repeats (total length of the contig repeated with
100% identity).
contigs as the nodes of the graph, out of which 13,842 contigs are exact repeats (total length of the contig repeated with
100% identity).
To begin, our evaluation involves assessing the method across various steps of the pipeline. Specifically, we examine the
outcomes relative to the baseline, the results produced by the GNN (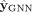 ), the outputs generated by RF (
), the outputs generated by RF (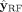 ), and finally, after the fine-tuning step, (
), and finally, after the fine-tuning step, (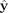 ). In this context, the term “baseline” refers to a straightforward heuristic used to classify the contigs. This heuristic
relies on Step 3 and labels nodes according to the following criteria:
). In this context, the term “baseline” refers to a straightforward heuristic used to classify the contigs. This heuristic
relies on Step 3 and labels nodes according to the following criteria: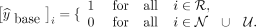 (6)
This approach allows us to test the effectiveness of sequencing features in node labeling in the absence of graph-based features.
(6)
This approach allows us to test the effectiveness of sequencing features in node labeling in the absence of graph-based features.
In Figure 4A, it is evident that the F1-score consistently rises throughout the pipeline, emphasizing the importance of each step in achieving optimal results. The baseline method exhibits high precision (98.3%) but low recall (40.9%), indicating appropriate node selection for determining pseudolabels but an inability to identify most repeats. This observation underscores that sequencing features alone are insufficient for detecting repeats. This limitation is modified by the GNN, which significantly boosts the recall to 68.6%, effectively identifying more repeats, which suggests that graph structure is significant in detecting the repeats. Subsequent application of the RF further amplifies this increase in recall to 80.2%. However, this enhanced recall comes at the cost of reduced precision compared to the baseline. To address this precision loss, the fine-tuning step effectively identifies outliers, leading to a precision increase from 72.6% at the output of the RF to 83.8% for the final estimation. In summary, our approach yields an 88.9% F1-score without any prior labels on the contigs, representing a substantial improvement of 31.2% over the baseline method.

Behavior of GraSSRep across different steps. (A) Progression of the method's performance throughout the different steps, highlighting the effectiveness of each step in improving repeat detection. We also test the impact of excluding the GNN embeddings and RF step applied to the augmented feature vectors. (B) High importance of GNN-generated embeddings in RF classification.
Moreover, we investigate the impact of the GNN and the embeddings it generates. To assess this, we perform two analyses. First,
we exclude Z from the feature matrix fed to the RF, resulting in 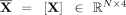 aiming to observe the method's performance only based on the initial graph-based features. As depicted in Figure 4A under “excluding GNN,” this exclusion leads to a decrease in all performance metrics. This decline suggests that embeddings
play a crucial role in enhancing the reliability of repeat detection. Second, we calculate the importance of the features
fed to the RF by averaging the impurity decrease from each feature across trees. The more a feature decreases the impurity,
the more important it is. These importance values are then plotted in Figure 4B. The plot indicates that all learned embeddings (labeled z1 through z16) exhibit high importance. This finding emphasizes
the utility of the embeddings generated by the GNN in improving the overall performance of the method. Further discussion
on the effect of the GNN can be found under “GNN effect” in the Supplemental Material. Moreover, by removing the intermediate RF step and directly applying the fine-tuning process to the GNN-generated labels,
we evaluate the effect of the RF step. As illustrated in Figure 4A under “excluding RF,” this omission also results in a decrease in all performance metrics, highlighting the essential role
of the RF in balancing the influence of initial features and the learned embeddings.
aiming to observe the method's performance only based on the initial graph-based features. As depicted in Figure 4A under “excluding GNN,” this exclusion leads to a decrease in all performance metrics. This decline suggests that embeddings
play a crucial role in enhancing the reliability of repeat detection. Second, we calculate the importance of the features
fed to the RF by averaging the impurity decrease from each feature across trees. The more a feature decreases the impurity,
the more important it is. These importance values are then plotted in Figure 4B. The plot indicates that all learned embeddings (labeled z1 through z16) exhibit high importance. This finding emphasizes
the utility of the embeddings generated by the GNN in improving the overall performance of the method. Further discussion
on the effect of the GNN can be found under “GNN effect” in the Supplemental Material. Moreover, by removing the intermediate RF step and directly applying the fine-tuning process to the GNN-generated labels,
we evaluate the effect of the RF step. As illustrated in Figure 4A under “excluding RF,” this omission also results in a decrease in all performance metrics, highlighting the essential role
of the RF in balancing the influence of initial features and the learned embeddings.
Additionally, we perform an ablation study on the percentile value p used to define the thresholds in Steps 3 and 5. The analysis in the section “Ablation study on the percentile value p” in the Supplemental Material reveals that our approach is robust to this hyperparameter, particularly within the range of 30 to 40, which corresponds to the range used in our experiments.
Lastly, if we replicate the analysis in Figure 4 with an alternative graph construction method, we observe that all outcomes align consistently as outlined under “Alternative graph construction” in the Supplemental Material. This illustrates the versatility of our tool, demonstrating its efficacy across diverse graph structures.
Comparison with existing repeat detection methods
We present a comprehensive comparison of our method with several existing repeat detection methods using contigs assembled from the reads downloaded from ENA (Shakya 2). The ground-truth labels are obtained in the same manner as described under “Experimental setup,” using the reference genomes from the Shakya 1 data set.
We consider five widely recognized methods for this comparison. Opera (Gao et al. 2011) and SOPRA (Dayarian et al. 2010) identify repetitive contigs by filtering out those with coverage 1.5 and 2.5 times higher than the average coverage of all
contigs, respectively, without considering any graph structure. Similarly, the MIP scaffolder (Salmela et al. 2011) utilizes both high coverage (more than 2.5 times the average) and a high degree (50 or more) within the assembly graph to
detect the repeats. However, as the degree of contigs in the graph provided by metaSPAdes typically does not reach 50, we
utilize an adaptive approach. In this alternative, we adjust the threshold from 50th to the 75th percentile of the degrees
observed in the graph. Additionally, Bambus2 (Koren et al. 2011) categorizes a contig as a repeat if the betweenness centrality, divided by the contig length, exceeds the upper bound of
the range within c standard deviations above the mean on this feature. Here, c represents a hyperparameter of this method, and the optimal outcome on our data set was achieved with  . Lastly, MetaCarvel (Ghurye et al. 2019) employs four more complex graph-based features alongside coverage in a two-step process. First, any contig with a high betweenness
centrality (three or more standard deviations plus the mean) on the assembly graph is marked as a repeat. Moreover, a contig
is identified as a repeat if it falls within the upper quartile for at least three of these features: mean coverage, degree,
ratio of skewed edges (based on coverage), and ratio of incident edges invalidated during the orientation phase of the contigs
(for details, see Ghurye et al. 2019). Notably, because we utilize a contig graph instead of a scaffold graph, we do not incorporate the latest feature and adjust
the flag threshold from three to two in the second step.
. Lastly, MetaCarvel (Ghurye et al. 2019) employs four more complex graph-based features alongside coverage in a two-step process. First, any contig with a high betweenness
centrality (three or more standard deviations plus the mean) on the assembly graph is marked as a repeat. Moreover, a contig
is identified as a repeat if it falls within the upper quartile for at least three of these features: mean coverage, degree,
ratio of skewed edges (based on coverage), and ratio of incident edges invalidated during the orientation phase of the contigs
(for details, see Ghurye et al. 2019). Notably, because we utilize a contig graph instead of a scaffold graph, we do not incorporate the latest feature and adjust
the flag threshold from three to two in the second step.
As illustrated in Figure 5, GraSSRep outperforms all other methods, particularly demonstrating superior capability in detecting repeats with a higher recall rate (66.3% vs. the next best alternative at 56.2%).
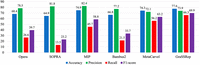
GraSSRep compared with the other repeat detection methods.
Thus far, we have focused on the practical unsupervised setting in which no repeat labels are available. For completeness, we now consider the case in which repeat labels for some contigs are available. This setting might arise, for example, if we have knowledge about specific organisms present in the metagenomic sample and their corresponding reference genomes are accessible. GraSSRep can seamlessly accommodate this case. In our pipeline, we can leverage this prior knowledge to substitute Step 3. Instead of pseudolabels, we employ the known node labels as our training set, leading to a semisupervised (instead of self-supervised) setting. Our analysis in the section “Incorporating prior knowledge” in the Supplemental Material shows that performance can be markedly improved in the case in which labels are available for a fraction of the contigs.
Discussion
We tackled the challenging task of detecting repetitive sequences (repeats) in metagenomics data when we only have access to paired-end reads. We introduced GraSSRep, a novel method that leverages the inherent structure of the assembly graph by employing GNNs to extract specific features for the contigs. Moreover, adopting a self-supervised learning framework, we generated noisy pseudolabels for a subset of the contigs, which were then used to train a graph-based classifier on the rest of the contigs.
Experimental studies using simulated data sets demonstrated the robustness of GraSSRep across diverse repeat characteristics and its resilience not only to repeat length but also to copy number variations. This ensures its applicability across various data sets and scenarios. Moreover, using synthetic data sets, we show the value of every step in our algorithm in enhancing repeat detection performance. This highlights the importance of each step and its role in achieving the best results. Furthermore, the GNN step effectively learns distinctive and important features for the repeat detection task based on the data set, thereby enhancing the pipeline's ability to detect more repeats using the graph structure.
Additionally, we observed performance gain compared to existing repeat detection tools. This superiority comes from the combined
value of incorporating learnable graph features (through the GNN) and considering a self-supervised framework. Notice that
even if we fix the embedding dimension at  , the graph features learned by the GNN depend on the specific data set under consideration. In this way, our trainable architecture
can distill the key graph features that characterize repeats in the specific metagenomic sample. This adaptive approach stands
in contrast to other methods, which often rely on fixed features. Moreover, because the RF is not pretrained but rather trained
based on the pseudolabels, different features may vary in importance based on context. In this way, our self-supervised framework
allows us to adapt to the metagenomic data at hand, and we do not have to worry about generalization issues of pretrained
models.
, the graph features learned by the GNN depend on the specific data set under consideration. In this way, our trainable architecture
can distill the key graph features that characterize repeats in the specific metagenomic sample. This adaptive approach stands
in contrast to other methods, which often rely on fixed features. Moreover, because the RF is not pretrained but rather trained
based on the pseudolabels, different features may vary in importance based on context. In this way, our self-supervised framework
allows us to adapt to the metagenomic data at hand, and we do not have to worry about generalization issues of pretrained
models.
One limitation of our work is its dependence on the initial pseudolabels. Specifically, in order to effectively generalize the labels from the initial training set to the other unlabeled nodes, we need sufficient samples in both repeat and nonrepeat sets of training contigs from a diverse set of organisms. However, this process can be hindered by unbalanced coverage across different organisms. When some organisms exhibit significantly higher coverage compared with the rest of the community, the contigs generated from these organisms tend to dominate the high percentile of coverage and are detected as repeats in Step 3. Consequently, our training set becomes biased toward a few organisms, impeding the detection of repeats of other organisms. To address this issue, we plan to develop a more systematic approach to training set selection in future work.
Furthermore, it is worth noting that although we selected the indicated reference genomes for the Shakya community for ground-truth detection and evaluation of our method, previous studies (Ondov et al. 2019) have identified additional reference genomes present in the community. Consequently, some repeats may be missing from the ground-truth set, as their reference genomes are not included in the community and thus not identified as true repeats.
A natural extension of our approach is its integration into widely used assemblers. This integration would replace their existing repeat detection modules with GraSSRep, yielding potential improvements in assembly quality. We also intend to apply our method to real data sets, particularly in environments like hot springs where widely accessible reference genomes are scarce. Lastly, the overall pipeline of GraSSRep can potentially address other problems in genomics in which graph structures can be used to identify specific genetic markers in the absence of prior knowledge. For instance, we intend to leverage our approach for the identification of transposable elements, which play important roles in eukaryotic/mammalian genomes.
Software availability
An implementation of GraSSRep, along with the code to reproduce our results, are available at GitHub (https://github.com/aliaaz99/GraSSRep) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Science Foundation under award EF-2126387.
Author contributions: A.A., A.B., T.J.T., and S.S. conceived and designed the study. A.A. and S.S. developed the methods and theory. A.A. and A.B. performed the experiments. A.A., T.J.T., and S.S. conducted the analyses. All authors analyzed and discussed the results. A.A. and S.S. drafted the initial manuscript, which was reviewed and edited by all authors. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279136.124.
- Received February 15, 2024.
- Accepted July 15, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








