
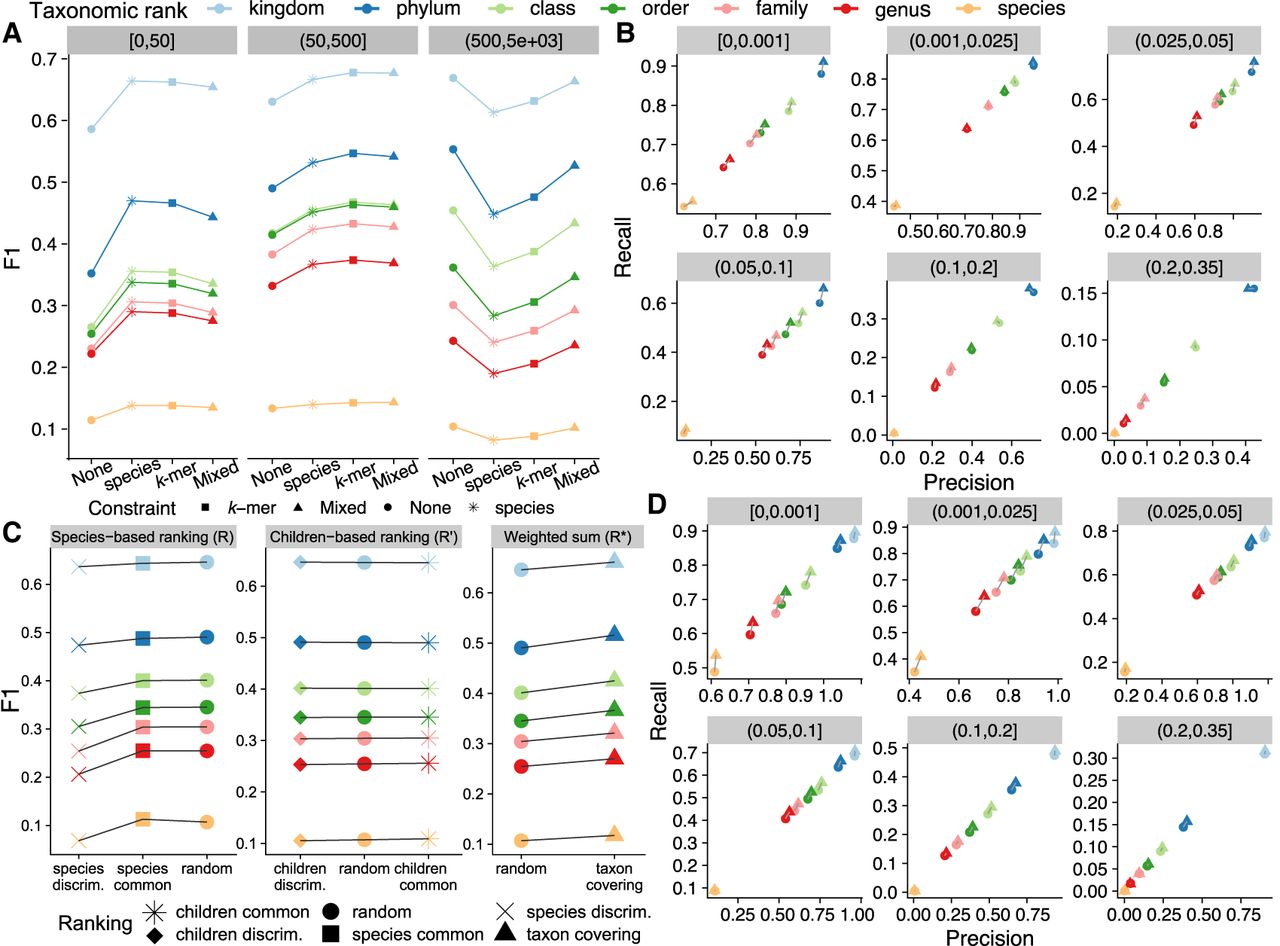
Evaluation of read classification performance of KRANK using different heuristics for k-mer selection. (A,B) Comparison of adaptive size-constraint approaches with random k-mer selection. None: enforcing no constraint, species: number of species constraint, k-mer: total k-mer count constraint, Mixed: no constraint for one table and k-mer count constraint for the other. (A) F1 accuracy, dividing queries (panels) into three bins based on the number of reference genomes in the phylum of the query. (B) Precision versus recall of None versus Mixed (default), dividing queries into bins of novelty (d*). (C,D) Evaluation of simple ranking strategies (discriminative, common, random) implemented using species counts R or children counts, R′ and our new weighted-sum, R* heuristic [Equation (11)]. See Supplemental Figure S2 for an illustration of functions. (C) F1 across all queries. (D) Precision versus recall of random and weighted-sum heuristic, dividing queries by bins of novelty (d*). In (A) and (C), x-axes are ordered by the average F1 score of all queries. All results use: w = 35, k = 32, l = 2, h = 12, and b = 16. d* is the distance to the closest reference estimated by Mash. Default KRANK uses mixed adaptive size and R* ranking.








