
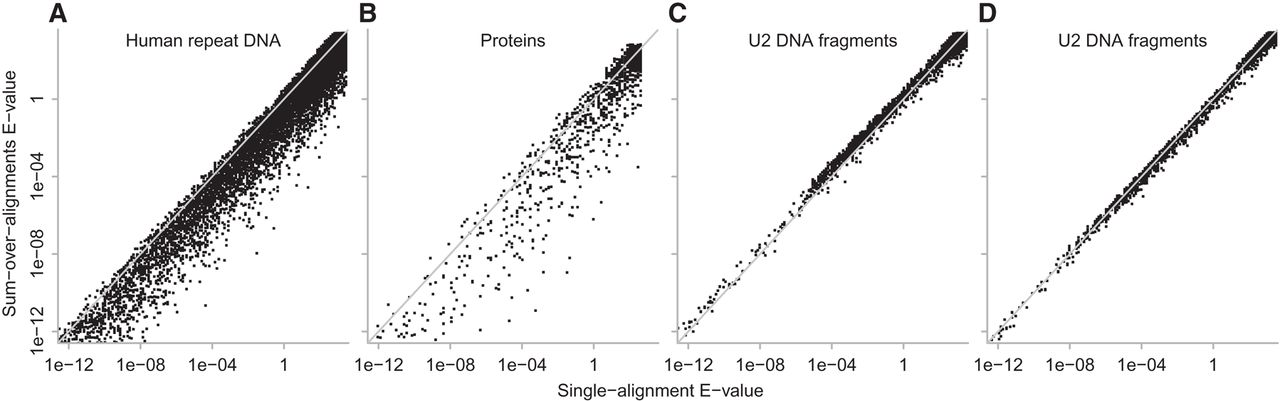
Figure 6.
E-values for identical alignments found by Algorithm 1 (horizontal axis) and Algorithm 2 (vertical axis). Each point is one alignment. The diagonal gray lines indicate equal E-values. These E-values were calculated with m = total length of all reference sequences (e.g., all A. aeolicus proteins) and n = total length of all query sequences (e.g., all P. fumarii proteins). For DNA, one query strand was searched against both reference strands, so m was multiplied by two. (A) Human repeat DNA; (B) proteins; (C) U2 DNA fragments; (D) U2 DNA fragments with fitted match/mismatch/gap probabilities.








