
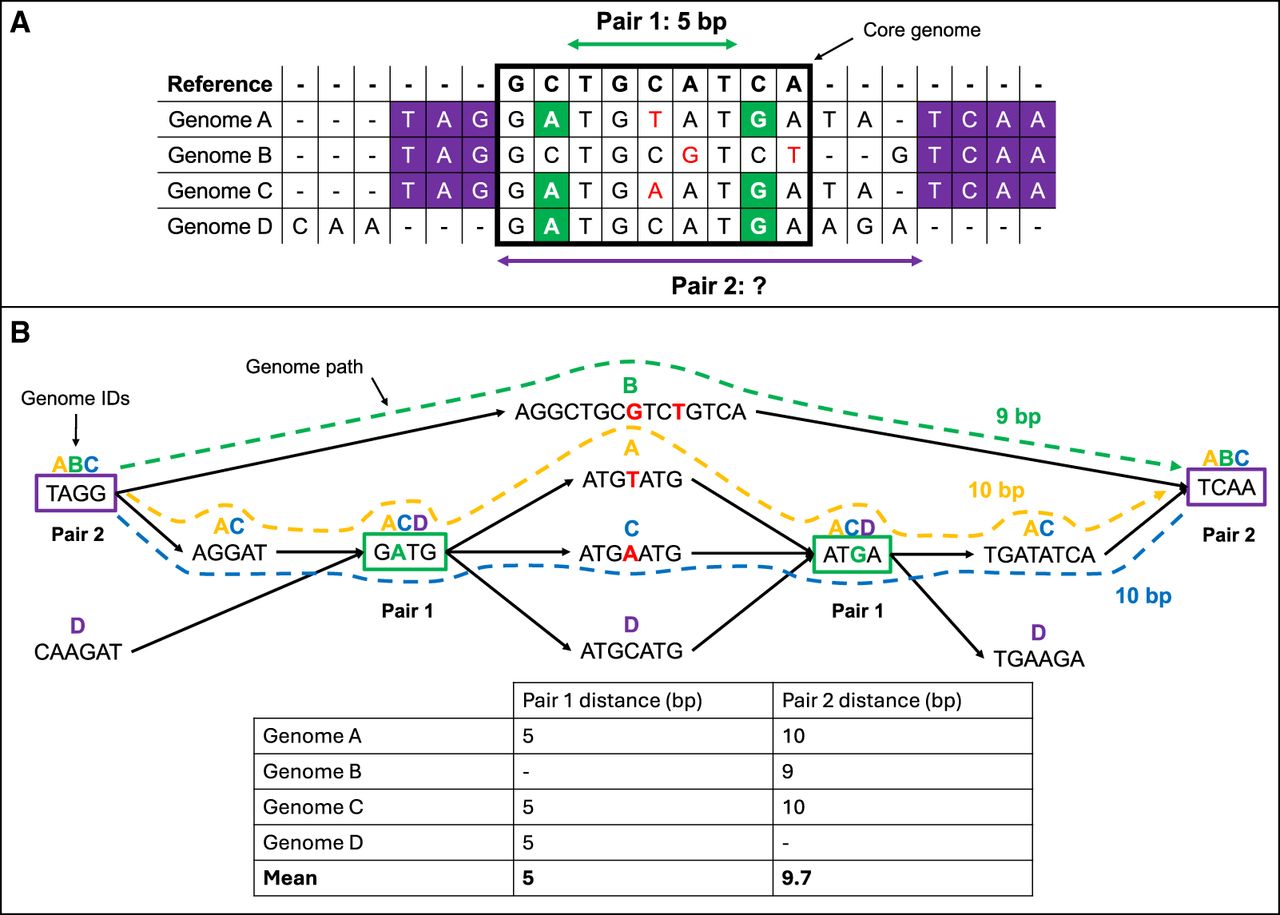
Comparison of MSA- and DBG-based GWES. (A) MSA-based GWES uses distances calculated from alignment of genomes to a reference. Only variants found within blocks of sequence shared with the reference, for example, within the core genome, can be detected and have distances calculated between them, exemplified by pair 1. Pair 2, which is variants found within the accessory genome, do not align to the reference, meaning the distance between them cannot be calculated and so is ignored from the mutual information (MI) calculation. (B) Using a DBG, distances can be calculated for pair 2 by traversing the graph for each genome containing this pair of variant, and then by calculating the mean distance between them. The DBG can also be used to calculate the distance for pair 1 using the same approach. Therefore, variant pairs in both the core and accessory genome can be identified.








