
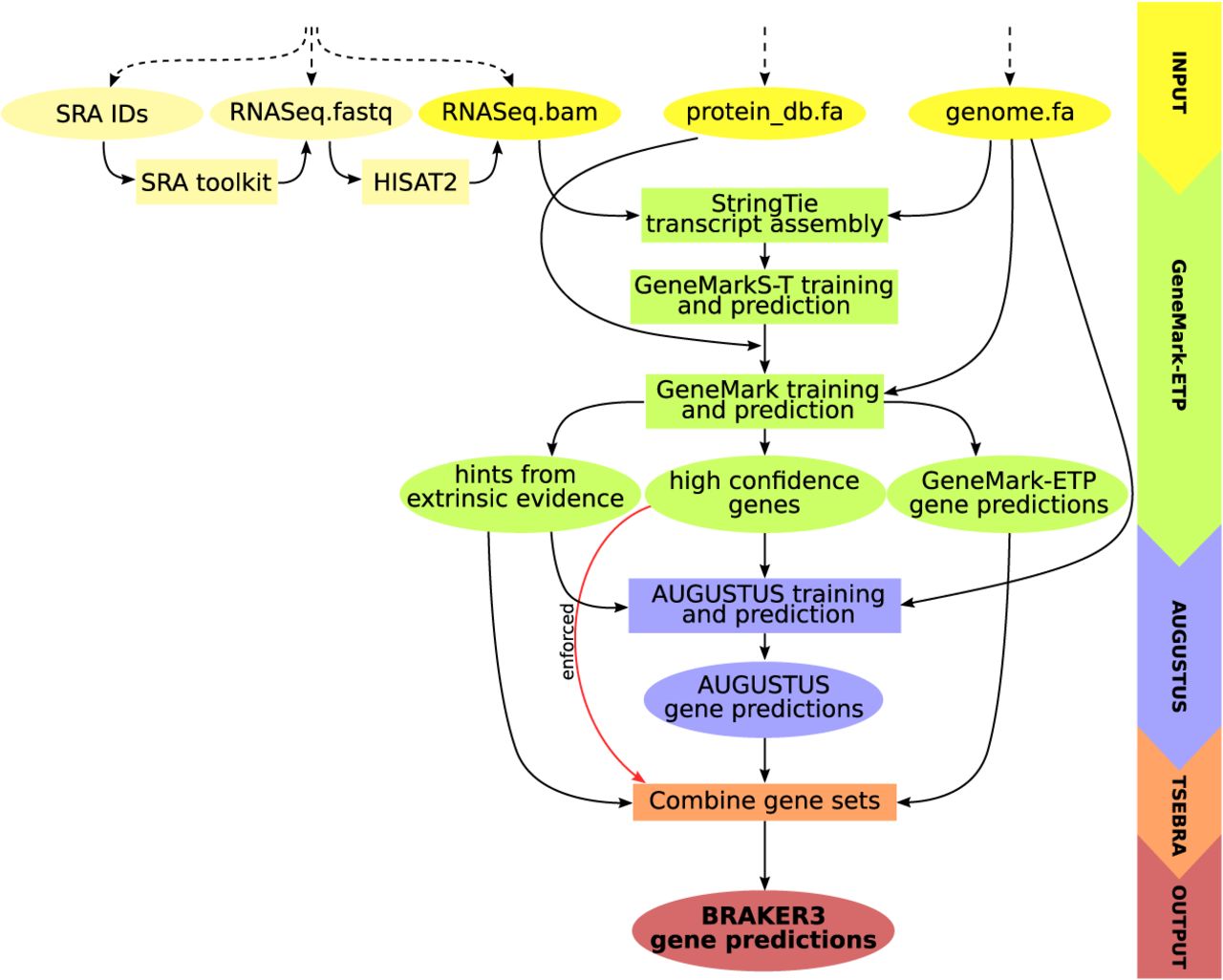
Figure 1.
Schematic view of the BRAKER3 pipeline. Required inputs are genomic sequences, short-read RNA-seq data, and a protein database. The RNA-seq data can be provided in three different forms: IDs of libraries available at the Sequence Read Archive (Leinonen et al. 2010), unaligned reads, or aligned reads. If library IDs are given, BRAKER3 downloads the raw RNA-seq reads using the SRA Toolkit (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software) and aligns them to the genome using HISAT2 (Kim et al. 2019). It is also possible to use a combination of these formats when using more than one library.








