
Abstract
Gastric cancer (GC) is the fifth most common cancer worldwide and is a heterogeneous disease. Among GC subtypes, the mesenchymal phenotype (Mes-like) is more invasive than the epithelial phenotype (Epi-like). Although gene expression of the epithelial-to-mesenchymal transition (EMT) has been studied, the regulatory landscape shaping this process is not fully understood. Here we use ATAC-seq and RNA-seq data from a compendium of GC cell lines and primary tumors to detect drivers of regulatory state changes and their transcriptional responses. Using the ATAC-seq data, we developed a machine learning approach to determine the transcription factors (TFs) regulating the subtypes of GC. We identified TFs driving the mesenchymal (RUNX2, ZEB1, SNAI2, AP-1 dimer) and the epithelial (GATA4, GATA6, KLF5, HNF4A, FOXA2, GRHL2) states in GC. We identified DNA copy number alterations associated with dysregulation of these TFs, specifically deletion of GATA4 and amplification of MAPK9. Comparisons with bulk and single-cell RNA-seq data sets identified activation toward fibroblast-like epigenomic and expression signatures in Mes-like GC. The activation of this mesenchymal fibrotic program is associated with differentially accessible DNA cis-regulatory elements flanking upregulated mesenchymal genes. These findings establish a map of TF activity in GC and highlight the role of copy number driven alterations in shaping epigenomic regulatory programs as potential drivers of GC heterogeneity and progression.
Gastric cancer (GC) is the fifth most common cancer worldwide and is more prevalent in Asia as well as among Asian Americans in the United States (Lui et al. 2014; Taylor et al. 2015; Ajani et al. 2017). Despite advances in early diagnosis and treatments, the overall 5-yr survival rate is still ∼30% (Badgwell et al. 2017). GC is a heterogeneous disease. A landmark study of The Cancer Genome Atlas (TCGA) proposed four molecular subtypes of GC, including Epstein–Barr virus (EBV)-associated, microsatellite instable (MSI), genomically stable (GS), and chromosomal instability (CIN) (Sohn et al. 2017). Histological subtyping using the Lauren classification is also associated with therapeutic recommendations (Berlth et al. 2014).
To determine personalized therapeutic responses to different treatments, previous studies have shown how treatment responses correlate with the gene expression profiles of GC (Tan et al. 2011), as well as the activity and mutation status of frequently mutated genes such as CDH1 (previously known as E-cadherin), TP53, and ARID1A (Park et al. 2016; Luo et al. 2018; Xu et al. 2023). Among such classifications are the mesenchymal (Mes) and epithelial (Epi) phenotypes, in which the former confers more invasive disease (Oh et al. 2018). The process by which epithelial cells gain a mesenchymal phenotype is called the epithelial-to-mesenchymal transition (EMT) (Brabletz et al. 2018). Expression profiles of Mes GC and their correlation with response to therapeutic agents such as 5-fluorouracil and PI3K inhibitors have been investigated (Tan et al. 2011; Lei et al. 2013; Oh et al. 2018). In addition, the activity of transcription factors (TFs) regulating normal stomach and GC (GATA4, GATA6, KLF5, HNF4A, and EBF1) has been implicated in GC invasiveness using TF and histone ChIP-seq (Chia et al. 2015; Xing et al. 2020; Sheng et al. 2021). In Mes GC, TEAD1 was identified as a major TF regulator (Ho et al. 2023) using genome-wide chromatin accessibility assays (ATAC-seq). However, there is currently a lack of systematic and unbiased approaches to uncover the complete range of TFs and epigenomic alterations underlying GC heterogeneity.
Here, we expand upon this earlier work by generating the first systematic map of GC TF activity derived from an unbiased machine learning approach applied to ATAC-seq data sets from a broad compendium of GC cell lines and primary TCGA tumors (Bass et al. 2014; Corces et al. 2018), coupled with RNA-seq analysis for interpretation of the altered regulatory pathways. Our analysis aims to infer the complete set of TFs whose activity explains the heterogeneity of chromatin landscapes across GC and to connect TF activity through enhancers to transcriptional activation of specific gene responses. We further investigate mutational and copy number (CN) alterations as genomic aberrations underlying the dysregulation of these key TFs. Finally, we relate GC cell line subtypes to other tissues using models trained on accessibility and single-cell transcriptional profiles.
Results
Sequence-based machine learning model of ATAC-seq profiles identifies distinct regulatory programs driving GC heterogeneity
To understand differences in epigenomic state profiles in gastric-cancer derived cell lines, we generated ATAC-seq data from a collection of 25 cell lines (Ho et al. 2023). Principal component analysis (PCA) of variation in the activity of distal peaks (Methods) showed significant heterogeneity in chromatin accessibility across the cell lines, forming three clusters (Fig. 1A) with similar genomic accessibility profiles. We used a machine learning approach, gkm-SVM (Ghandi et al. 2014a; Lee et al. 2015; Beer et al. 2020), to identify the sequence features (transcription factor binding sites [TFBSs]) driving these variations in chromatin accessibility. We chose gkm-SVM because of its combination of predictive accuracy and interpretability (Ghandi et al. 2014a, 2016; Lee et al. 2015; Lee 2016; Mo et al. 2016; Beer 2017; Kreimer et al. 2017; Gate et al. 2018; Shigaki et al. 2019; Beer et al. 2020; Yan et al. 2021) and because of its advantages relative to DNNs when training on smaller numbers of differentially active peaks (about 2000). For each cell line, we trained a gkm-SVM model on the top 10,000 distal ATAC-seq peaks against random genomic sequences, following our standard pipeline (Beer et al. 2020). On each training, following the default training method in the gkmSVM-R package (Ghandi et al. 2016), the AUROC was evaluated on fivefold cross-validation (CV), and the reported AUROC is the average of five CV test sets. The median CV AUROC is 0.925. All cross-fold validation sets produce similar sets of features and have very similar weight vectors. Because this test set AUROC is high, there is little evidence of overfitting. Also, following the method of Ghandi et al. (2016) for feature interpretation but not for quantitative assessment, we use the gapped k-mer weight vector when training on all the data to reduce variability in the inferred features. Each gkm-SVM model summarizes the TFBS required to classify the ATAC-seq peaks in a vector of gapped k-mer weights that can predict the accessibility of the sequences. We clustered the gkm-SVM weight vector from each cell line (Fig. 1B) and found nearly identical clusters to Figure 1A; thus, the similar genomic accessibility profiles are explained by activation of shared TFs.
Variation in the chromatin accessibility profiles across GC cell lines is consistent with differential TF activity inferred from sequence-based machine learning. (A) PCA of ATAC-seq detects variable accessibility clustered roughly into three groups of GC cell lines (Mes-like, Intermediate, Epi-like). (B) gkm-SVM training produces similar weight vectors in the Mes-like, Intermediate, and Epi-like GC cell lines, resulting from the activation of similar sets of TFs. (C) Training gkm-SVM on differentially active peaks detects RUNX, AP-1, and TEAD activation in Mes-like peaks and detects KLF, GATA, GRHL, FOXA, and HNF4A activation in Epi-like peaks. Similar TF activation is detected when training on a primary GC tumor (TCGA-STAD) versus normal stomach. ZEB is a transcriptional repressor, and the absence of its motif from open-chromatin regions shows its activity and, hence, the flipped sign (red color) for ZEB activity score. (D) TFBS PWM logos. (E) gkm-SVM inferred activity (dot size) of these TFs across all samples detects common patterns of activation but additional heterogeneity within each group. (F) ChIP-seq validation experiments show that RUNX2 and AP-1 bind to Mes-high distal peaks and do not bind to Epi-high peaks in LPS141, consistent with the machine learning predictions of RUNX and AP-1 activity. On the other hand, GATA4, GATA6, and KLF5 bind to Epi-high peaks and do not bind to Mes-high peaks in AGS. AGS is one of the Epi-like GC cell lines, and LPS141 is a mesenchymal liposarcoma cell line with a very similar transcriptional profile to Mes-like GC cell lines (see Supplemental Fig. S6).
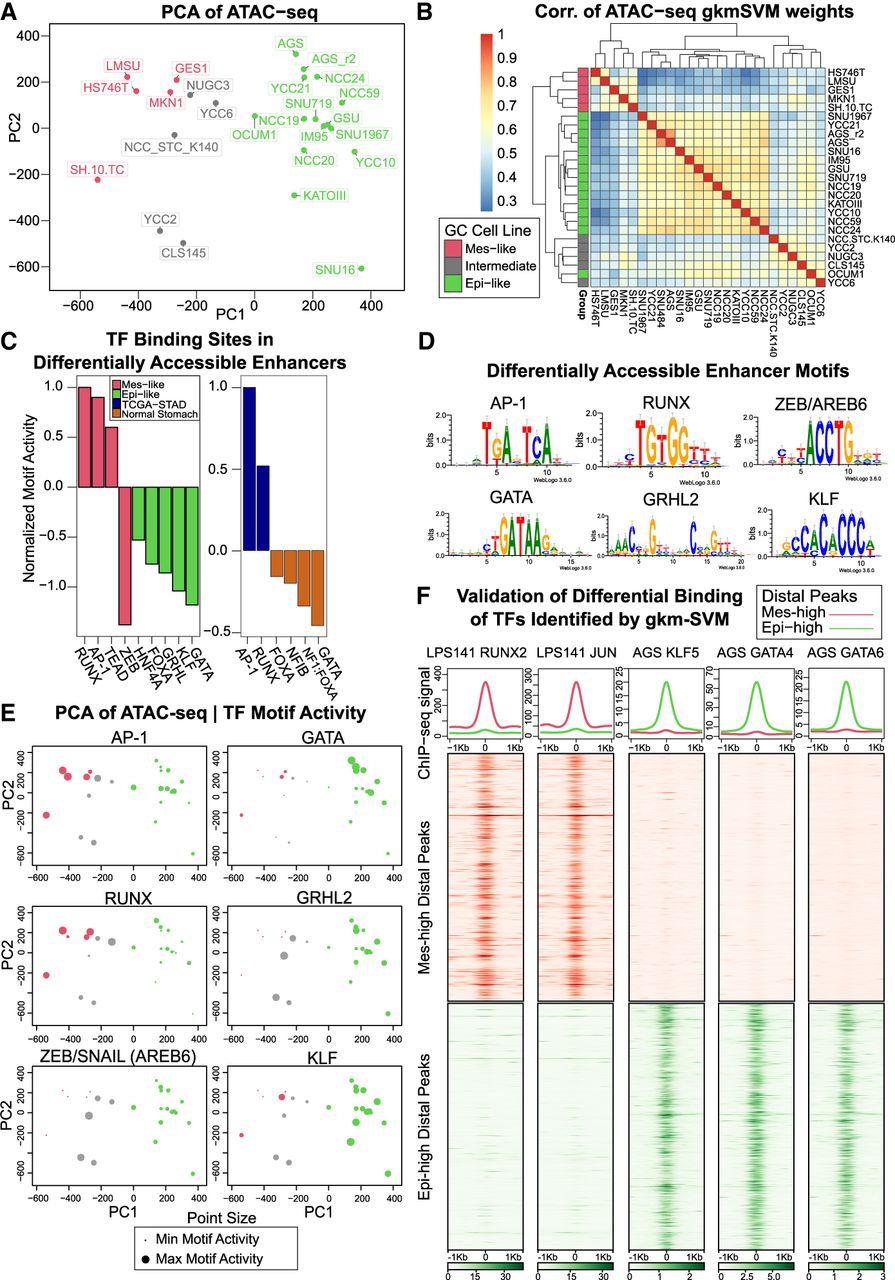
Based on these differentially active TFs and their resultant differentially expressed (DE) target genes, which largely align with the EMT and mesenchymal-related pathways (see the RNA-seq Analysis section below), we label these three sets of GC cell lines as epithelial-like (Epi-like), Intermediate, and mesenchymal-like (Mes-like). To isolate the TFs driving most of the variability between these sets of cell lines, we next trained gkm-SVM on the most differentially accessible peaks when averaged over the Epi-like and Mes-like cell lines, with Mes-like peaks as the positive set, and Epi-like peaks as the negative set (Supplemental Figs. S1, S2). Again, this generated a highly predictive sequence model, and the gkm-SVM weights with the largest difference between the Epi-like and Mes-like cell lines were associated with the differential activity of TFs binding the specific motifs: RUNX, AP-1 (FOS/JUN), TEAD, ZEB, HNF4A, FOXA, GRHL, GATA, and KLF (Fig. 1C; Supplemental Fig. S3). Because ZEB1 and ZEB2 are known transcriptional repressors (Aghdassi et al. 2012; Byles et al. 2012), ZEB activity is indicated by a negative motif score. To assess if these TFs also drive variation in tumors, we next trained gkm-SVM on differential peaks in normal stomach versus primary stomach tumor samples (The Cancer Genome Atlas for Stomach Adenocarcinoma [TCGA-STAD]) (Fig. 1C; Supplemental Figs. S4, S5). The TFs detected largely overlap with those in the cell lines, indicating that these TFs also drive variation in the regulatory state between normal stomach tissue and GC tumors. Although there is significant heterogeneity among the tumor samples and although only a fraction of the cells in a tumor are likely in a mesenchymal state, we find significant evidence that the Mes-like regulatory program identified in the cell lines is activated in the STAD samples relative to normal stomach. Seventy percent of the high-quality TCGA-STAD ATAC-seq samples detect RUNX, and 70% detect AP-1 (Methods) (Supplemental Fig. S5). Finally, we trained gkm-SVM on differentially accessible peaks in each pair of cell lines (300 pairs; see Methods) to detect the full set of TFs explaining the ATAC-seq variation across the cell line collection. RUNX and AP-1 (and lack of ZEB) were strong TFBS signals driving accessibility in Mes-like cell lines, whereas GRHL, GATA, and KLF TFBS were strongly associated with the most Epi-like samples (Fig. 1D,E). In summary, this ATAC-seq analysis of open chromatin identifies RUNX, AP-1, ZEB, GRHL, GATA, and KLF as novel regulatory drivers of the Epi-like and Mes-like transcriptional states in GC. These predictions from gkm-SVM are validated by experiments showing strong RUNX2 and JUN (which binds AP-1) binding at Mes-like enhancers from ChIP-seq in LPS141 (Bevill et al. 2023) and strong KLF5, GATA4, and GATA6 binding at Epi-like enhancers from ChIP-seq generated in AGS (Fig. 1F; Supplemental Fig. S6; Chia et al. 2015; Liu et al. 2020).
Regulatory and expression signatures of EMT and advanced-versus-early GC explain the differences between GC groups
We used RNA-seq data from the 25 GC cell lines to understand how the differential TF activity inferred from the ATAC-seq data affects gene expression. The three groups of GC cell lines, previously clustered based on their chromatin accessibility, were recapitulated using the expression of tissue-specific genes (Methods) (Fig. 2A; Supplemental Fig. S7), showing that the variation in RNA-seq is concordant with that of ATAC-seq.
RNA-seq expression of GC cell lines is concordant with EMT and advanced-versus-early GC expression signatures and is consistent with TF activity inferred from chromatin accessibility. (A) PCA of gene expression profiles is consistent with PCA from ATAC-seq. (B) Combined PCA of GC cell lines with TCGA-STAD (tumor) and TCGA-normal shows shared variation between Mes-like versus Epi-like and tumor versus normal. (C,D) The genes explaining the variation (top 100 PCA gene weights) in the direction of PC1 (A) and PC2 (B) both are most enriched for genes highly expressed in EMT and advanced-versus-early GC. (E) Examples of marker gene expression for EMT in cancer (higher in Mes, red; higher in Epi, green), with likelihood ratio test, FDR < 0.05, and |log2FC| > 2. (F) Differentially expressed (DE) TFs between the Mes-like and Epi-like GC cell lines, with a likelihood ratio test FDR < 0.05 and |log2FC| > 2. (G) Survival analysis based on the expression of Mes and Epi DE TFs in the ACRG cohort.
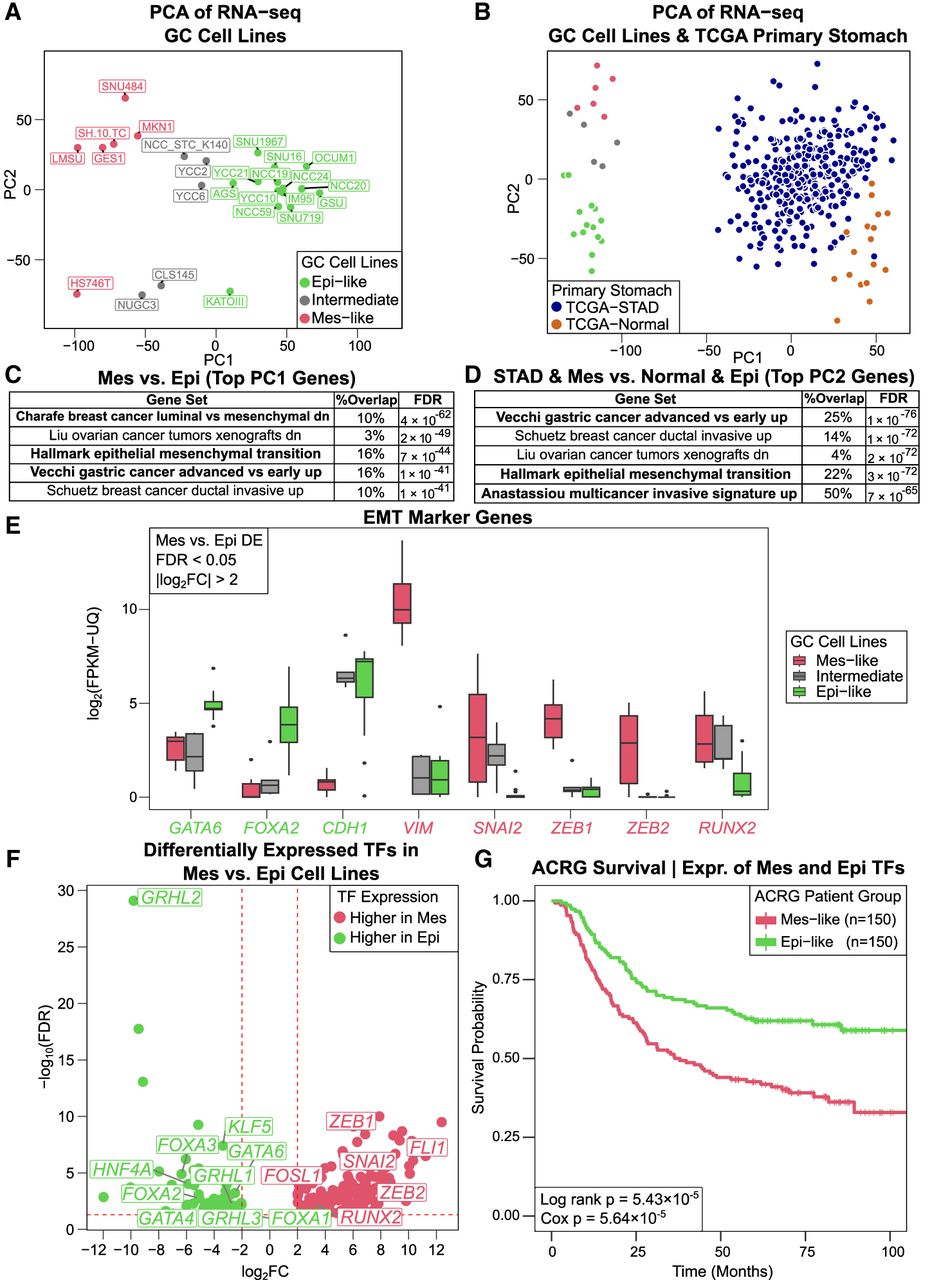
We next compare gene expression in GC cell lines with that of primary stomach tissue from TCGA-STAD and TCGA normal adjacent tissue (Methods). The combined PCA with cell lines and TCGA shows that there are inherent differences between the stomach-derived cell lines and stomach primary tissue on the first principal component (PC1) (Fig. 2B), as noted in previous studies (Aran et al. 2015; Yu et al. 2019). PC2 shows TCGA-Normal samples are more aligned with Epi-like GC cell lines, whereas TCGA-STAD cancer samples are closer to Mes-like cell lines (Supplemental Fig. S8).
To associate these expression differences with biological processes, we performed gene set enrichment analysis using PCA gene weights and gene set enrichment analysis (GSEA) (Mootha et al. 2003; Subramanian et al. 2005; Liberzon et al. 2011, 2015). The PC1 of Figure 2A was strongly associated with Mes and EMT phenotypes, advanced-versus-early GC, and cancer invasiveness (hypergeometric FDR < 10−40) (Fig. 2C; Charafe-Jauffret et al. 2006; Schuetz et al. 2006; Vecchi et al. 2007; Liberzon et al. 2015). EMT is also the top hit when using expression changes between Mes-like and Epi-like clusters (Supplemental Figs. S9, S10) instead of PC1 weights. We discovered a similar pattern of enrichment comparing TCGA-STAD and normal tissues with the cell lines (Fig. 2D, which is gene set enrichment analysis on PC2 of Fig. 2B). Taken together, this shows that the biological differences between the Mes-like and Epi-like clusters of the GC cell lines overlap with the EMT and invasiveness phenotypes. Because of these associations, we label the red group of cell lines in Figures 1A and 2A “Mes-like” for their mesenchymal expression signature (EMT); the green group is labeled “Epi-like” for their more normal Epi expression profile; and the gray group of cell lines is labeled “Intermediate.” The Intermediate set lies along the continuum between Epi-like and Mes-like but is distinguishable from the Mes-like set through the stronger activity of ZEB and GRHL motifs and is distinguishable from the Epi-set by the lack of GATA activity (see Fig. 1E). Our ATAC-seq analysis allows us to identify TF regulators with these EMT and invasiveness gene expression signatures.
TF regulators detected in ATAC-seq are markers of EMT and invasiveness and are DE in Mes-versus-Epi GC cell lines
To further explore the connection between regulatory states and cancer invasiveness, we generated a set of 2679 DE genes between the Mes-like and Epi-like cell lines (Methods). These DE genes include the regulator TFs implicated from ATAC-seq, as well as many genes previously implicated in Epi GC or EMT (Supplemental Tables S1, S2). We observed that TFs such as GATA4, GATA6, KLF5, HNF4A, GRHL2, FOXA1, FOXA2, and FOXA3 in the Epi-like cell lines, as well as SNAI2, ZEB1, FOSL1 (AP-1), and RUNX2 in Mes-like, not only have a significantly higher expression (Fig. 2E,F; Supplemental Table S1) but also have differentially accessible DNA-binding sites in Mes-like versus Epi-like GC cell line ATAC-seq (Fig. 1C). Further, we assessed the expression of these DE TFs in an independent patient cohort from the Asian Cancer Research Group (ACRG; Methods) (Fig. 2G; Cristescu et al. 2015), as well as in TCGA-STAD (Supplemental Fig. S11), and found that differential TF expression is associated with overall survival, suggesting that activation of these regulator TFs has both biological and disease prognostic utility.
Many of the DE genes have been previously implicated in cancer. In the Epi-like upregulated group, KLF5 and HNF4A along with GATA4 and GATA6 have been shown to promote GC (Chia et al. 2015). GRHL2 plays an anti-EMT role in GC (Luo et al. 2019; Sheng et al. 2021) through TGFB signaling, whereas GATA6 suppresses EMT by promoting the expression of FOXA2 and CDH1 while decreasing the activity of SNAI2, TWIST1, ZEB1, and VIM in pancreatic cancer (Martinelli et al. 2017). TFs in the Mes-like upregulated group, ZEB1, RUNX2, and SNAI2, are known regulators of EMT (Niu et al. 2012; Brabletz et al. 2018; Roche 2018). Additionally, FOSL1 dimerizes with JUN family members to bind the AP-1 motif and drives EMT by upregulating ZEB1/2 and SNAI2 (Bakiri et al. 2015; Guneri-Sozeri et al. 2023). The combined differential expression of these TF genes and the identification of their TF binding sites as the largest signals in predictive sequence modeling of the chromatin accessibility profiles strongly suggest that the variability in activation of this small set of TFs is responsible for driving the EMT program. Although this finding is in the context of GC, the fact that LPS141 arrived at a similar transcriptional state and has RUNX2 and JUN binding at the Mes-like enhancers suggests that this may be a more general mechanism of activating EMT across other tumor types (Supplemental Fig. S6).
Mutations and CN alterations are associated with distinct regulatory states in GC
We next sought to identify possible mechanisms driving the variation in regulatory state across the GC cell lines. To understand how the activity of TFs in Mes-like and Epi-like cell lines correlated with known genetic variation in GC, we examined TF gene expression in TCGA-STAD patients. CDH1, a recurrently mutated GC driver gene (Supplemental Table S3; Fodde 2002; Lei et al. 2013; Luo et al. 2018; Xu et al. 2023), was downregulated in Mes-like GC cell lines compared with the Epi-like. We examined the expression of DE TFs in CDH1-mutated versus CDH1-WT TCGA-STAD patients (Methods) (Fig. 3A; Supplemental Fig. S12). CDH1-mutated TCGA-STAD tumors showed upregulation of the Mes-like TFs. Conversely, the CDH1-WT tumors displayed upregulation of Epi-like TFs (Fig. 3A). This result indicates that CDH1 mutations in GC correlate with upregulation of key EMT-promoting TFs and downregulation of EMT-inhibiting TFs.
Mutations and copy number (CN) variants are associated with distinct Mes-like and Epi-like TF activation in GC. (A) Nonsynonymous CDH1 mutation status separates TFs with differential expression between Mes-like and Epi-like cell lines. Shown is TCGA-STAD expression in tumors with and without a CDH1 nonsynonymous mutation (t-test P < 0.05 and |log2FC| > 0.25). (B) Human TFs ranked in order of DNA CN alteration rate in TCGA-STAD primary tumors. (C) Chromatin accessibility landscape and TF binding of ZEB1 and SMAD4 in the GATA6 locus. GATA6_E1 and GATA6_E2 are putative Epi-like and normal stomach enhancers, where GATA6 binds in GATA6 ChIP-seq in AGS GC cell line. (D) GATA4 has reduced CN in non-Epi cell lines, and MAPK9 has amplified CN in non-Epi cell lines. Average CN differences between Epi GC cell lines and non-Epi (Mes and Intermediate) GC cell lines are shown for protein-coding genes whose expression and CN correlate. CN values are obtained by DNA sequencing, in which “−2” marks deep DNA deletion, “0” shows a normal diploid genome, and “+2” is for deep DNA amplification in the gene locus. Mann–Whitney U test P < 0.05. (E) CN variation is consistent with altered GATA4 and MAPK9 expression across samples. r is the Pearson's correlation between CN scores and gene expression.
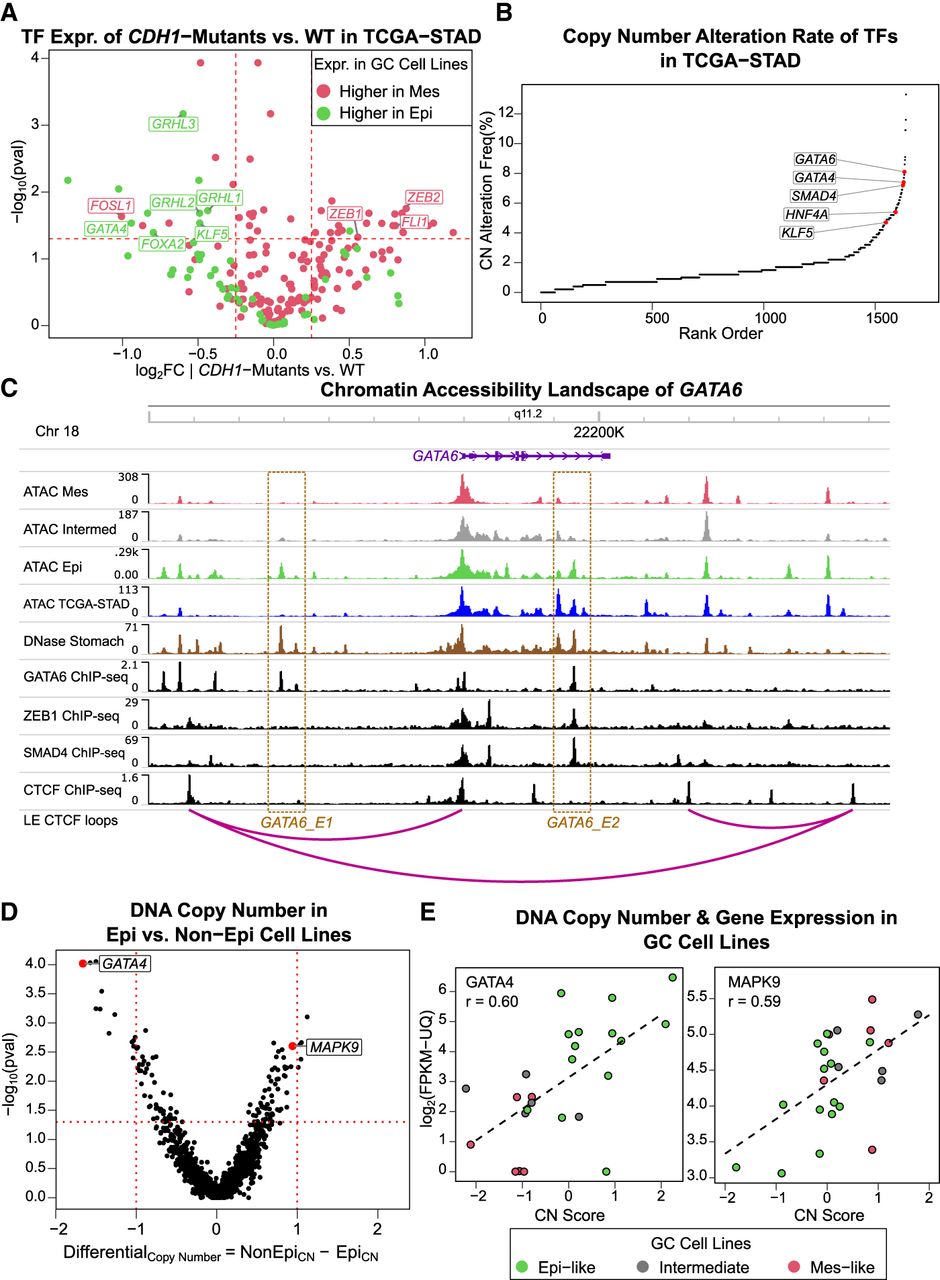
In addition, the TFs GATA4, GATA6, SMAD4, KLF5, and HNF4A are among the TFs with the highest rates of DNA CN alterations in TCGA-STAD patients (Fig. 3B; Supplemental Fig. S13). GATA4, GATA6, and SMAD4 are known to be important in endoderm and stomach development (Li et al. 2019), and SMAD4 has been identified as a stomach cancer driver gene (Martínez-Jiménez et al. 2020). GATA4 and GATA6 have lower expression in Mes-like GC cell lines (Fig. 2F), consistent with gkm-SVM analysis (Fig. 1E). ATAC-seq profiles for Mes-like, Intermediate, and Epi-like GC cell lines also identify two distal peaks (GATA6_E1 and GATA6_E2) in the GATA6 locus (Fig. 3C) whose accessibility correlates with GATA6 expression (Supplemental Figs. S14, S15). GATA6_E1 is upstream of GATA6 and bound by GATA6 in AGS, and GATA6_E2 is intronic and is also bound by GATA6 in AGS and by SMAD4 and ZEB1 in HepG2 cells. The low ATAC signal in Mes-like cell lines at GATA6_E2 is consistent with the upregulation of the repressor ZEB1 and downregulation of GATA6. In further support of their regulatory role, both GATA6_E1 and GATA6_E2 are contained in loop extrusion (LE) model–predicted CTCF loops containing the GATA6 promoter (Xi and Beer 2021; Luo et al. 2023). Overall, the upregulation of the Epi regulator GATA6 is consistent with the Mes-like and Epi-like ATAC-seq profiles through GATA6_E1 and GATA6_E2.
Reduced GATA and increased AP-1 regulatory activity in GC cell lines are accompanied by DNA CN alterations
The gkm-SVM-inferred differential GATA activity can be attributed to either GATA6 or GATA4 dysregulation, and both GATA4 and GATA6 have lower expression in Mes-like GC (Fig. 2F). To identify potential driving mechanisms, we compared CN changes in Epi-like cell lines versus non-Epi cell lines (Mes and Intermediate), using DNA sequencing (Methods). On average, GATA4 has a higher CN in Epi-like cell lines (Fig. 3D), which correlates with lower expression of GATA4 in Mes-like cell lines (r = 0.56) (Fig. 3E). Although <1% of TCGA-STAD patients have a mutation in GATA4 according to cBioPortal (Gao et al. 2013), GATA4 CN alterations are more frequent in TCGA-STAD (Fig. 3B). Further, an interval of Chr 8p containing GATA4 is frequently deleted in human epithelial cancers (Cai and Sablina 2016). Thus, GATA CN alterations are a potential mechanism for GATA4 expression changes (Fig. 2F) and GATA activity in GC cell lines and primary tumors (Fig. 1C). We observed an opposite pattern for MAPK9 kinase, which is amplified in the Mes-like cell lines compared with Epi-like (Fig. 3D,E). MAPK and JNK family members are known to be regulators of AP-1 (Karin et al. 1997), and MAPK9 amplification is associated with higher expression of AP-1 complex members (FOSL1) (Fig. 2F) and higher AP-1 transcriptional activity in Mes-like cell lines and primary stomach cancer (Fig. 1C). Taken together, we see evidence for DNA CN changes in TFs and signaling pathway components as potential drivers of transcriptional dysregulation in GC cell lines.
Mes-like GC cell lines have expression and regulatory signatures similar to fibroblasts, whereas Epi-like GC cell lines retain gastrointestinal signatures
Because we detected the activation of nonstomach TFs among many GC cell lines, we next sought to characterize their cell states by comparing them with known cell types. We first compared the GC cell line expression profiles to known normal cell types by calculating their correlation with Genotype-Tissue Expression (GTEx) RNA-seq data (Methods) (Fig. 4A; Lonsdale et al. 2013). Mes-like GC cell line expression was more similar to fibroblasts (r = 0.44) than to stomach (r = 0.29), whereas Epi-like and Intermediate were most similar to stomach and esophagus mucosa, respectively (Fig. 4A). We further confirmed that the gene expression in Mes-like GC cell lines is more similar to fibroblast-derived cells than to stomach in two additional independent RNA data sets from ENCODE and TCGA-STAD (Methods) (Fig. 4B; The ENCODE Project Consortium 2012; Luo et al. 2020). Although TCGA-Normal is more similar to normal stomach than TCGA-STAD, the similarity of TCGA-STAD to fibroblast is weaker than the Mes-like cell lines (Fig. 4B).
In both expression and chromatin accessibility, Mes-like GC cell lines are more similar to fibroblasts, and Epi-like GC cell lines are more similar to stomach. (A) Mes-like expression is most similar to GTEx fibroblasts. Correlation of 54 healthy GTEx gene expression profiles for each group of GC cell lines in about 11,300 tissue-specific genes. (B) Correlation of ENCODE stomach and fibroblast gene expression profiles with average gene expression for each group of GC cell lines. HT1080 is a fibrosarcoma cell line. (C) Mes-like chromatin accessibility is also most similar to an ENCODE fibroblast, and the TCGA-STAD and Epi-like GC cell lines are most similar to ENCODE stomach. We measured the similarity of chromatin accessibility by correlation of gkm-SVM weight vectors trained on distal enhancers of each sample. ELR is a fibroblast-derived cell line. (D) Correlation of GC cell line gene expression profiles with a healthy ENCODE lung fibroblast and a healthy stomach sample. (E) Correlation of GC cell line chromatin accessibility profiles (gkm-SVM weights trained on cell line ATAC-seq) with healthy ENCODE lung fibroblasts and stomach (gkm-SVM trained using DNase-seq).
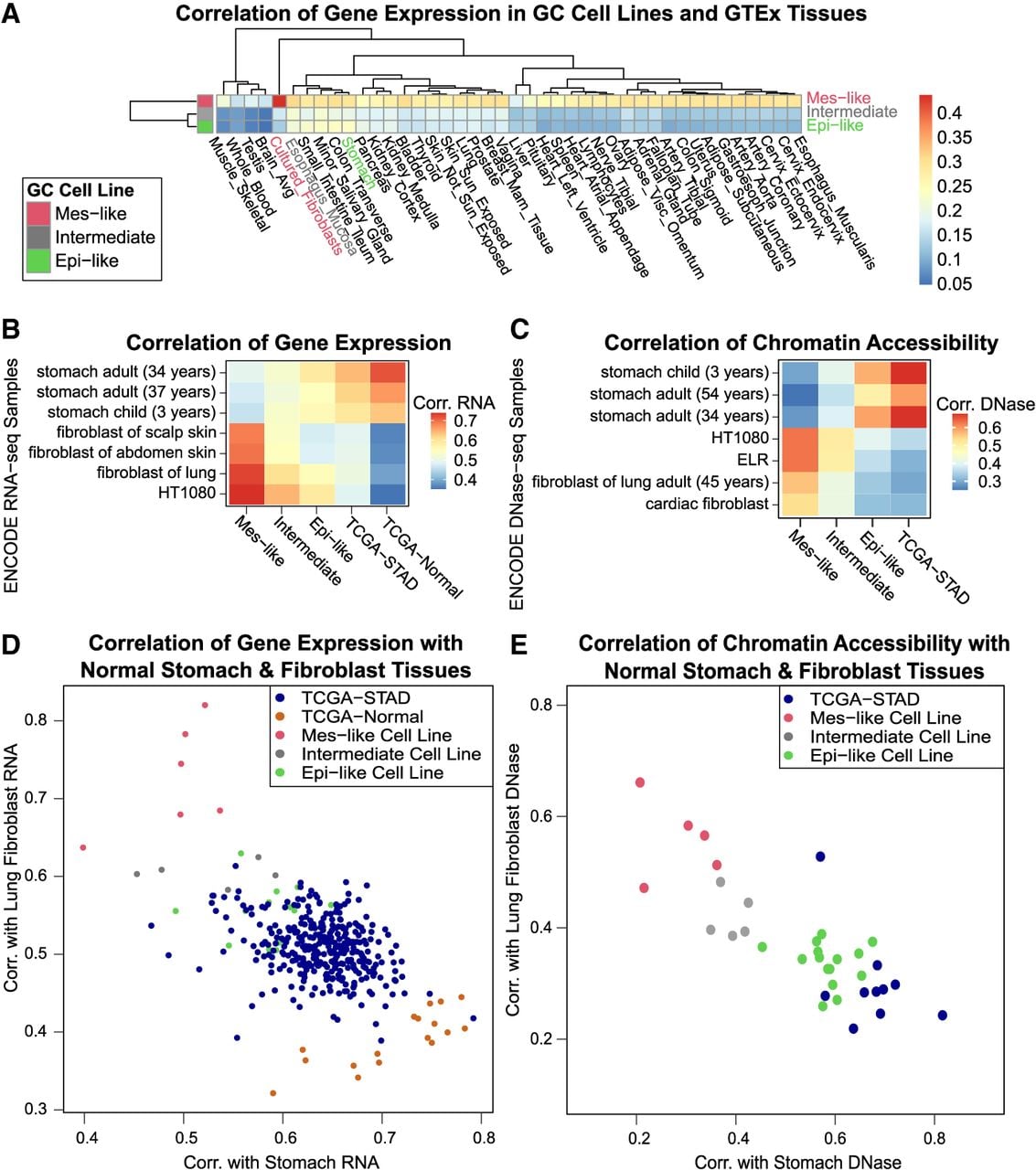
We extended these comparisons to DNase-seq data and found consistent similarities between the chromatin accessibility of the Mes-like GC cell lines with ENCODE DNase-seq of fibroblast (Fig. 4C). We trained gkm-SVM sequence models on open chromatin regions for each GC cell line, TCGA-STAD, ENCODE fibroblast, and stomach tissues and compared their weight vectors (Methods). The chromatin accessibility of Mes-like GC is highly correlated with the fibroblast-derived cell lines HT1080 and ELR, as well as primary fibroblast tissue from ENCODE, as opposed to primary stomach. In contrast, the Epi-like cell lines and TCGA-STAD ATAC-seq were more correlated with the primary stomach.
In addition to these average correlations of expression and chromatin accessibility, we now show that differential TF activity among GC cell lines positions them along a continuum of cell states between normal stomach and fibroblast. Scatter plots of correlation to stomach and fibroblast (Fig. 4D,E; Supplemental Figs. S16, S17) show that TCGA-Normal had the highest correlation with ENCODE normal stomach and the lowest with fibroblast, whereas TCGA-STAD tumors have a higher correlation with fibroblast and a lower correlation with stomach (Fig. 4D). The Mes-like GC cell lines have a higher similarity to fibroblast than the primary tumors, correlations being as high as 0.8. Consistent with the RNA-seq profiles, gkm-SVM models trained on chromatin accessibility profiles also show that Mes-like cell line accessibility is more similar to fibroblast accessibility than normal stomach tissue (Fig. 4E; Supplemental Figs. S18, S19). Although it is clear that GC cell lines are becoming more similar to fibroblasts by the activation of fibroblast TFs, the bulk TCGA-STAD profiles may be moving toward a fibroblast cell state either by interaction with stroma through fibrosis or by the presence of cancer-associated fibroblasts (CAFs) in the tumor microenvironment (Kalluri and Zeisberg 2006; Piersma et al. 2020).
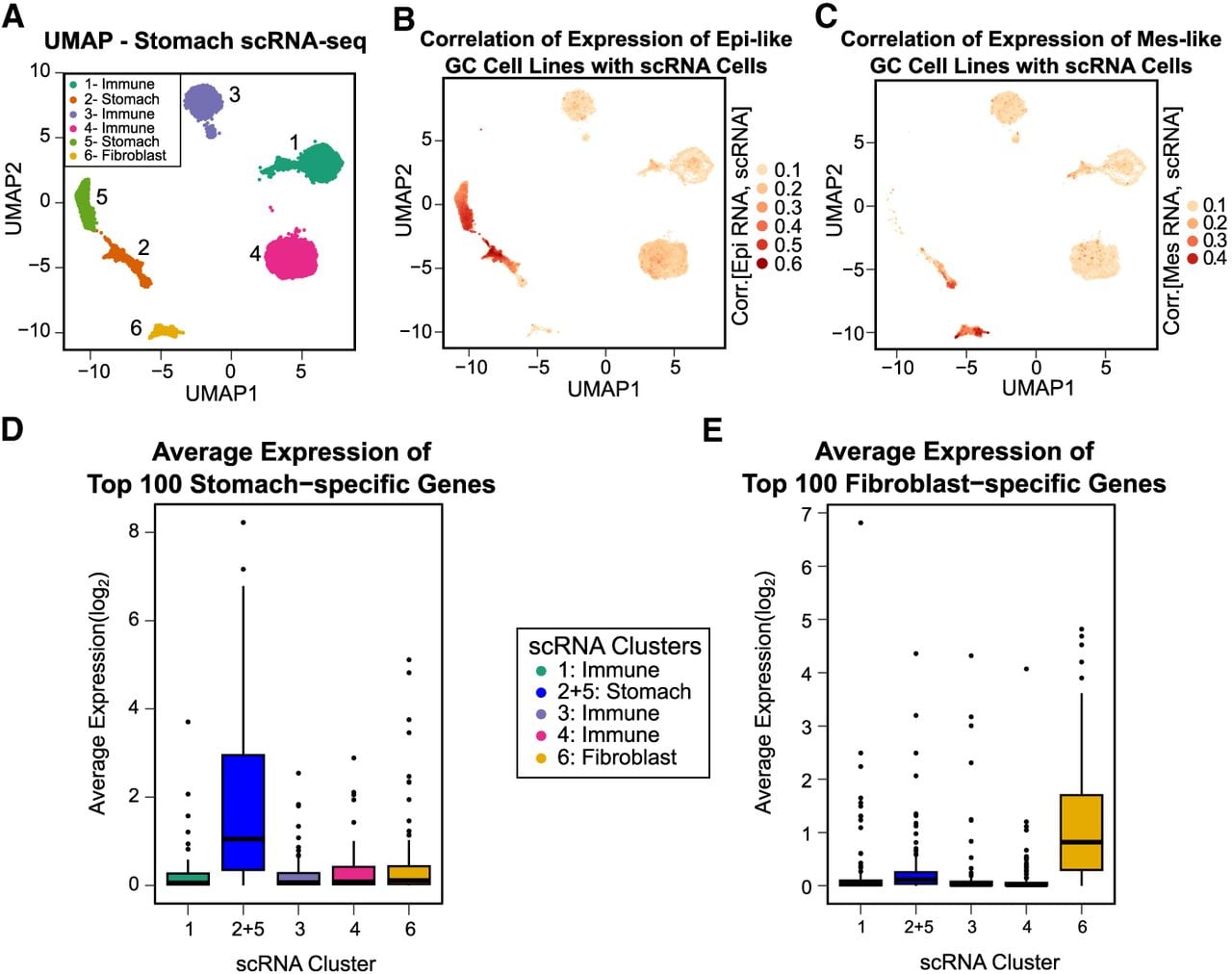
We next addressed cell line similarity at the single-cell level using stomach cancer scRNA-seq data from Kim et al. (2022) (Methods). We identified six distinct populations of cells using UMAP (Fig. 5A; Supplemental Fig. S20; McInnes et al. 2020), and based on marker gene expression (Methods) (Supplemental Table S4), we will identify clusters 2 and 5 as gastrointestinal and cluster 6 as fibroblast. We compared the expression of GC cell lines to these single-cell clusters and found high similarity between stomach clusters 2 and 5 with Epi-like GC cell lines (Fig. 5B) and high similarity between Mes-like GC cell lines and fibroblast cluster 6 (Fig. 5C). Expression of stomach- and fibroblast-specific genes from GTEx RNA-seq confirms our association of these clusters with these cell types (Methods) (Fig. 5D,E).
Mes-like GC is most similar in expression profile to the single-cell fibroblast population of both tumor and normal stomach tissue. (A) UMAP of scRNA-seq of gastric tumor and adjacent normal stomach tissue from Kim et al. (2022). Six cell populations are detected; populations 2 and 5 are stomach, and 6 is fibroblast. (B) Epi-like cell line gene expression profiles are most highly correlated with the stomach clusters 2 and 5. (C) Mes-like gene expression profiles are most highly correlated with the fibroblast cluster, cluster 6. (D) Highly expressed stomach-specific genes are expressed only in stomach clusters 2 and 5, combined owing to similarity (t-test P < 10−8 for cluster 2 + 5 vs. other clusters). (E) Highly expressed fibroblast-specific genes are expressed only in fibroblast cluster 6 (t-test P < 10−9 for cluster 6 vs. other clusters).
Fibrotic gene expression in GC cell lines is driven by activation of flanking enhancers
Although we observed similarity of ATAC-seq and RNA-seq profiles across Mes-like and Epi-like cell lines (Figs. 1A, 2A), we next show that their differential expression is consistent with direct induction by enhancers flanking the Mes-like and Epi-like genes. We calculated the average ATAC-seq signal in all distal ATAC-seq peaks within 50 kb of a transcription start site (TSS) of DE genes between Epi-like or Mes-like GC cell lines. The accessibility of peaks flanking genes more highly expressed in Epi-like cell lines is 1.73 times higher in Epi-like cell lines compared with Mes-like, and the accessibility of peaks flanking genes more highly expressed in Mes-like cell lines is 1.60 times higher in Mes-like cell lines compared with Epi-like ones (Fig. 6A,B), providing a direct connection between altered chromatin state and expression responses.
Mes and Epi upregulated genes are flanked by Mes-active and Epi-active distal enhancers. (A) Highly expressed genes in Epi-like GC cell lines are flanked by distal peaks with increased accessibility in Epi-like cell lines. (B) Highly expressed genes in Mes-like GC cell lines are flanked by distal peaks with increased accessibility in Mes-like cell lines. All peaks within 50 kb of TSS of DE genes were averaged. Many of the upregulated genes in Mes cell lines are members of the TGFB/SMAD and FGF pathways, and their expression is shown in C (Mann–Whitney U test P < 0.05). (D) In the COL1A1 locus, COL1A1_E1 and COL1A1_E2 are putative distal enhancers with increased ATAC-seq signal in Mes-like GC cell lines and TCGA-STAD relative to Epi-like and normal stomach, consistent with higher COL1A1 expression in Mes-like GC cell lines and TCGA STAD. ChIP-seq experiments in the mesenchymal liposarcoma cell line LPS141 identified AP-1 and RUNX2 binding in COL1A1_E1 and COL1A1_E2. (E) Similarly, in the FGF5 locus, FGF5_E is a putative distal enhancer with increased ATAC-seq signal in Mes-like GC cell lines and TCGA-STAD relative to Epi-like and normal stomach, consistent with higher FGF5 expression in Mes-like GC cell lines and TCGA STAD. FGF5_E is a RUNX2 and AP-1 binding site in LPS141. FGF5_E, COL1A1_E1, and COL1A1_E2 are contained in CTCF loops enclosing the target gene promoter.
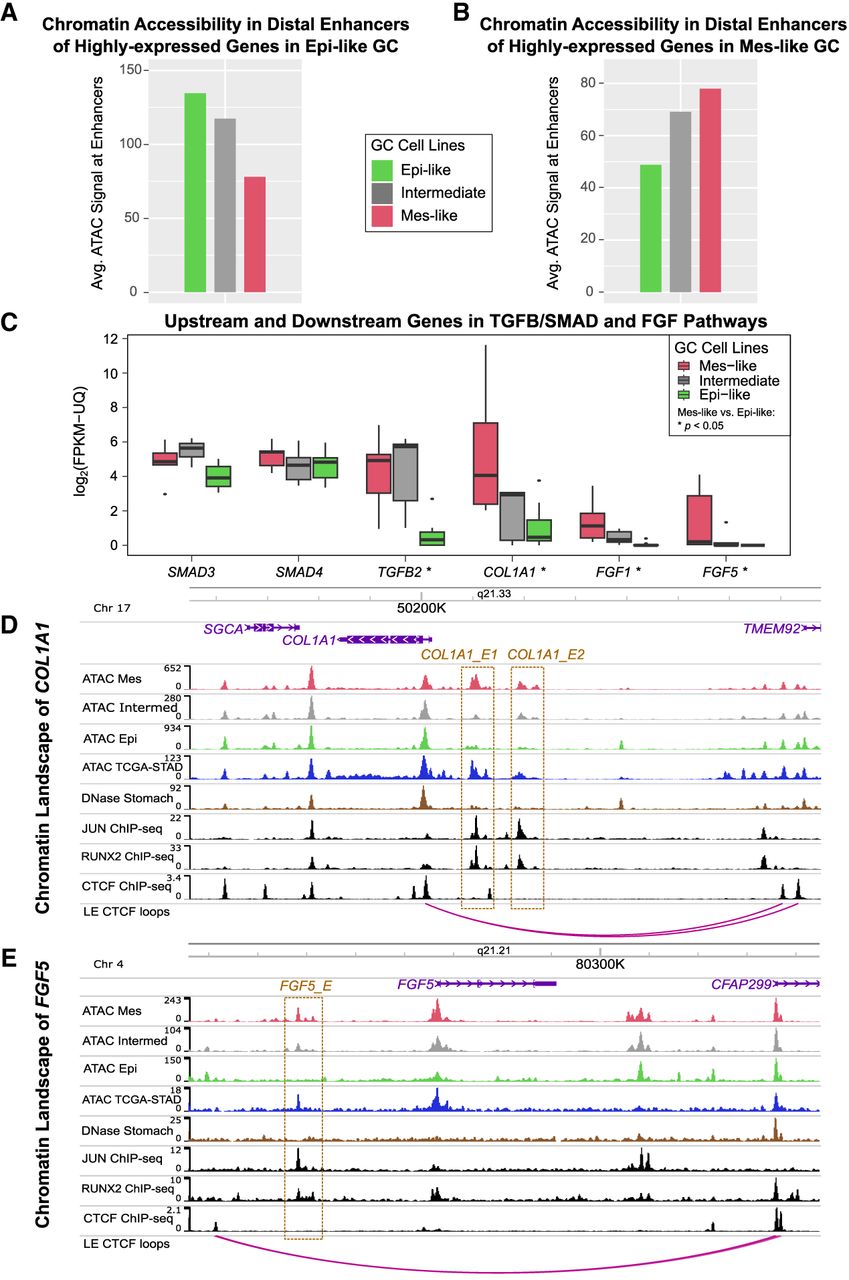
Among the genes upregulated in Mes-like cell lines are the fibrotic gene FGF5 and many collagen genes downstream from the TGFB/SMAD pathway (Verrecchia et al. 2001; Shin et al. 2019; Xie et al. 2020). Their role in gastrointestinal cancer (Luo et al. 2019) and cancer-associated fibroblasts (Bordignon et al. 2019) has been well established. Inhibition of FGF5 decreases proliferation and metastasis in hepatocellular carcinoma (Fang et al. 2015). COL1A1 has a similar effect on tumor behavior and proliferation (Nissen et al. 2019). FGF5, COL1A2, COL6A3, COL5A1, COL12A1, COL1A1, and COL6A2 are upregulated in Mes-like GC and have 1.5-fold to sixfold higher accessibility in flanking ATAC peaks. The members of the fibrotic pathways TGFB2, COL1A1, FGF1, and FGF5 are DE (Mann–Whitney U test P < 0.05) (Fig. 6C) in Mes-like and Epi-like GC.
To understand the regulatory mechanism for the differential expression of COL1A1 and FGF5, we compared the chromatin accessibility of their flanking cis-regulatory regions (Fig. 6D,E). For COL1A1, we found the upstream distal peaks COL1A1_E1 and COL1A1_E2 differentially active in Mes-like and Epi-like GC cell lines and in primary TCGA-STAD tumors compared with normal stomach (Fig. 6D). They are both located within a LE model–predicted CTCF loop (Xi and Beer 2021). Similarly, FGF5 is flanked by an upstream distal peak (FGF5_E) (Fig. 6E), which is active in primary TCGA-STAD cancer and inactive in healthy stomach tissue. Across cell lines, the expression of COL1A1 is highly correlated with the activity of the peaks COL1A1_E1 and COL1A1_E2 (r = 0.64 and 0.6) (Supplemental Figs. S21, S22). Similarly, the relatively higher expression of FGF5 in three of the five Mes-like cell lines is correlated with the activity of the distal peak (FGF5_E; r = 0.75) (Supplemental Fig. S23). COL1A1_E1, COL1A1_E2, and FGF5_E are all bound by RUNX2 and JUN in LPS141 (Bevill et al. 2023). Therefore, COL1A1 and FGF5 are dysregulated in both the Mes-like GC cell lines and stomach cancer tissue, consistent with the increased chromatin accessibility of newly identified flanking distal peaks, which are likely acting as enhancers.
Mes-like GC cell lines have a distinct regulatory landscape compared to fibroblasts
Because the Mes-like cell lines are epigenomically and transcriptionally similar to fibroblasts, we next sought to verify that they are not simply normal fibroblasts, or possibly CAFs (Sahai et al. 2020). Normal fibroblasts would be distinguishable by lower mutation or CN alteration rates. However, we found that the Epi-like, Intermediate, and Mes-like GC cell lines had indistinguishable mutation rates in the coding regions (t-test P > 0.12; Methods) (Fig. 7A). Additionally, we calculated the average rate of CN alterations in each cell line (Methods) (Fig. 7B), and the three groups of cell lines had similar rates. Although GES1 is derived from normal stomach epithelium, it was transformed with SV40 virus containing the T antigens, which downregulate TP53 and appear to have mimicked the high mutation burden of the other cell lines, which were not transformed with SV40 and do not have any ATAC-seq reads mapping to SV40. Because of these mutation and CN profiles, the Mes-like cell lines are clearly distinct from normal fibroblasts.
Despite some similarity in gene expression and enhancer activity, Mes-like GC and fibroblast have distinct TF responses. All groups of cell lines have similar levels of somatic mutations (A) and CN alterations (B). (C) PCA of chromatin accessibility of GC cell lines, stomach, and fibroblasts shows that although all groups are distinct, there is an axis along which differences in accessibility between stomach and fibroblasts are shared between the Epi-like and Mes-like cell lines. Epi-like cell lines are more similar to stomach, and Mes-like cell lines are more similar to but still distinct from, primary fibroblasts. (D) Mes-like versus fibroblast accessibility differences are driven by differential activity of a small set of TFs. (E) Rank plot of the average TF motif weights in all pairwise comparisons between Mes-like cell lines and ENCODE fibroblast samples. (F) Differential expression of TFs between the stomach and fibroblast shows that Mes-like cell lines retain high expression of stomach-specific TFs and do not upregulate all fibroblast TFs (especially TWIST2) to the same degree. Ranking of DE TFs between Mes-like and ENCODE-fibroblast (FDR < 0.01) in normal GTEx stomach (x-axis) and normal GTEx fibroblast profiles (y-axis) are shown. Some of the DE TFs upregulated in Mes-like GC (FOXA1, KLF5, etc.) have a higher expression (lower ranking) in GTEx normal stomach, whereas TWIST2 has a higher expression in ENCODE fibroblast and a higher expression (lower ranking) in GTEx fibroblast. (G) Inferred TF motif activities are supported by differential expression of stomach TFs and fibroblast genes in Mes-like GC, normal ENCODE-fibroblast, and ENCODE-stomach (**) FDR < 0.01, (*) FDR < 0.05, likelihood ratio test.
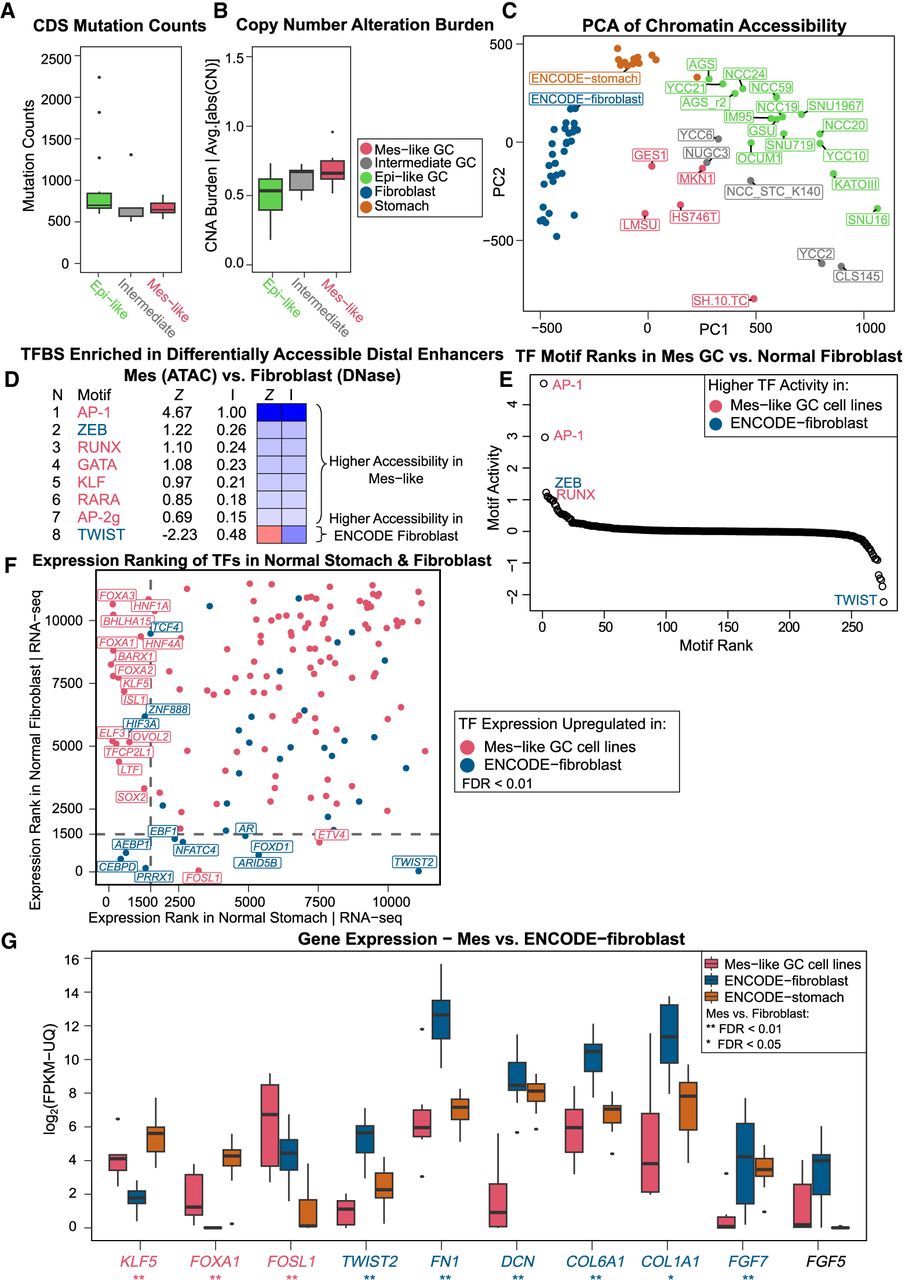
The Mes-like cell lines are also distinct from normal fibroblasts in their chromatin accessibility profiles. PCA analysis on distal open-chromatin regions in GC cell lines, normal fibroblast, and stomach ENCODE DNase-seq, showed differences between each group (Methods) (Fig. 7C, analysis similar to that in Fig. 1A). The variation between Epi-like, Intermediate, and Mes-like GC cell lines is aligned with an axis that also explains most of the variance between stomach and fibroblast accessibility profiles. In addition, gkm-SVM sequence models trained on differentially accessible ATAC distal peaks between Mes-like GC and normal fibroblasts highlight that TFs binding the AP-1, RUNX, GATA, RARA, and KLF motifs have higher activity in Mes-like GC, whereas fibroblasts have higher accessibility explained by TWIST and ZEB DNA-binding sites (Fig. 7D). By aggregating motif activity scores following pairwise comparisons between each Mes-like cell line and each ENCODE fibroblast, we rediscovered AP-1 and RUNX to be active in Mes-like GC as opposed to TWIST and ZEB being more active in normal fibroblasts (Fig. 7E).
Although the expression profiles of Mes-like GC cell lines are similar to fibroblasts, there are also systematic expression differences consistent with the above-noted chromatin accessibility changes. TFs with higher expression levels in normal stomach such as KLF5, FOXA1, and HNF4A were among the TFs having a higher expression in Mes-like GC compared with normal ENCODE fibroblasts (FDR < 0.01, |log2FC| > 2; Methods) (Fig. 7F). The differential activity of KLF, TWIST, and AP-1 discussed in the motif analysis above is consistent with the upregulation of KLF5 and FOSL1 in Mes-like GC compared with the higher expression of TWIST2 in normal ENCODE fibroblast (Fig. 7F). In addition, fibroblast marker genes such as FN1, DCN, COL6A1, COL1A1, and FGF7 have higher expression in ENCODE fibroblasts compared with Mes-like GC (Fig. 7G). Taken together, we see that despite the similar chromatin accessibility and gene expression signatures between Mes-like GC and fibroblast tissues, the epigenomic state of Mes-like GC can be clearly distinguished from fibroblasts by the activity of AP-1, RUNX, and the stomach motif KLF, as well as the lower activity of TWIST and ZEB compared with fibroblasts.
Discussion
We provide the first systematic map of GC TF activity inferred from an unbiased machine learning approach applied to ATAC-seq and RNA-seq data. Our sequence-based machine learning analysis revealed that most of the variation among the GC cell lines is driven by a fibrotic versus epithelial regulatory program differentially activated by a small set of TFs, most of which have not previously been directly associated with EMT in GC. We used a panel of GC-derived cell lines in combination with the TCGA stomach-adenocarcinoma (TCGA-STAD) cohort to identify heterogeneous regulatory programs in GC and their concomitant transcriptional responses. The inferred cell line regulatory programs explained much of the variation among TCGA-STAD samples. Our direct analysis of GC cell lines allowed for the isolation of cancer cell regulatory programs without noise derived from intra-tumor heterogeneity and variation in tumor immune infiltration.
Using ATAC-seq and computational sequence models, along with RNA-seq and scRNA-seq, we identified the largely novel set of TFs involved in Mes-like GC (RUNX2, SNAI2, ZEB1, AP-1 dimer) as opposed to the less-invasive Epi state (Epi-like GC) and its regulators (GATA4, GATA6, KLF5, HNF4A, FOXA2, GRHL2) (Fig. 8A). Mutation and DNA CN analysis identified genetic events associated with the activation of this regulatory program. The Mes-like transcriptional state was often associated with GATA4 DNA deletion and MAPK9 amplification (Fig. 8B). This suggests CN variation in TFs and signaling components can play a significant role as cancer driver mutations in addition to previously reported enhancer hijacking resulting from genomic structural rearrangement (Wang et al. 2021).
Summary of TFs driving GC epigenomic heterogeneity and EMT. (A) Differentially active TFs in Mes-like and Epi-like GC cell lines. (B) DNA CN changes in Mes-like and Epi-like GC cell lines. (C) Mes-like GC has a high correlation of expression and distal enhancer activity with fibroblasts, whereas Epi-like GC is more similar to the stomach. (D) Mes-like GC TF response and chromatin profile are different from that of fibroblasts, despite the shared patterns of expression and chromatin accessibility.
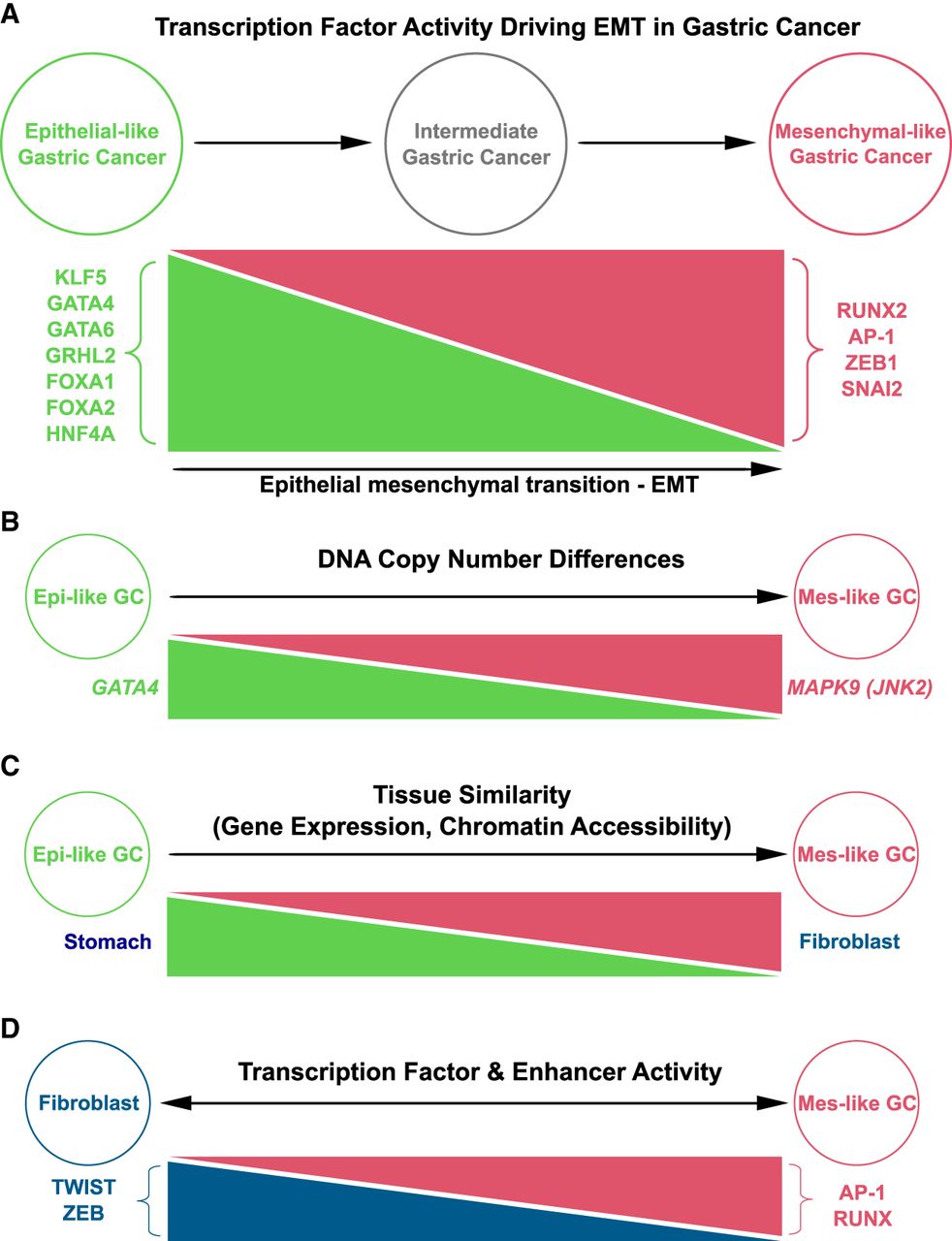
EMT in cancer is a complex process (Yang et al. 2020; Lovisa 2021), but we found that individual GC cell lines activate the gene expression signature associated with EMT to varying degrees. DNase-seq and RNA-seq from ENCODE and GTEx provided evidence for the existence of a fibrotic phenotype in Mes-like GC through EMT (Fig. 8C,D). Downstream fibroblast genes such as FGF5 and COL1A1 were found to be upregulated in both Mes-like GC and TCGA-STAD, and we identified flanking cis-regulatory enhancer elements with increased activity in Mes-like GC versus Epi-like GC, as well as TCGA-STAD compared with normal stomach. Survival analysis shows that activation of Mes-like TFs and their target genes is predictive of disease severity and suggests both prognostic and therapeutic utility.
Taken together, we identified altered regulatory programs in GC along with their distinct transcriptional responses driven by differential enhancer activity. We identify CN DNA alterations as genomic aberrations responsible for the dysregulation of these core TFs. Our findings suggest that activation of this small set of TFs driving the Mes-like GC regulatory program plays an important role in cancer progression, and highlight new biology and potential therapeutic opportunities in GC.
Methods
ATAC-seq data processing and gkm-SVM training
The raw GC cell line ATAC-seq data analyzed in this study was previously reported (Xing et al. 2020; Sheng et al. 2021; Ho et al. 2023; Xu et al. 2023) and uploaded to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) (see “Data access”). ATAC-seq paired reads were mapped to GRCh38 genome with Bowtie 2 version 2.2.5 (Langmead and Salzberg 2012). For gkm-SVM training, peaks were called by MACS2 (Zhang et al. 2008). Distal peaks were defined as 300-bp bins centered on the top 10,000 MACS2 peaks, after removing promoters (<2 kb of a TSS), and peaks open in >30% of ENCODE DNase-seq samples (mostly CTCF sites). Negative set genomic regions were selected randomly with matched GC and repeat content as described previously (Shigaki et al. 2019; Beer et al. 2020). Training positive peaks versus the negative sets generated robust models with a median CV AUROC of 0.925 (Supplemental Fig. S24). We used default settings and either the gkmSVM-R or lsgkm package (Ghandi et al. 2014a, 2016; Lee 2016). This training procedure was shown to generate gkm-SVM models with a strong performance in both MPRA experiments and predictions of variant impact (Beer 2017; Kreimer et al. 2017; Yan et al. 2021). For ENCODE DNase-seq normal stomach, we used ENCSR782SSS (Roadmap Epigenomics Consortium et al. 2015) downloaded from the ENCODE Portal (https://www.encodeproject.org), and for TCGA stomach tumor ATAC-seq, we used TCGA-BR-A4J6 (Supplemental Figs. S25, S26; Corces et al. 2018). ENCODE DNase-seq and TCGA STAD ATAC-seq (Corces et al. 2018; Shigaki et al. 2019) were processed in a similar manner, as described previously (Shigaki et al. 2019). We chose gkm-SVM for this task because of its robust performance on small (differential accessibility) data sets and because it has been shown that gapped k-mers are efficient representations of TFBS (Ghandi et al. 2014b; Yan et al. 2021) and protein motifs (Amanchy et al. 2011). Each gkm-SVM model generates a score function that can be specified as weights for each gapped k-mer (Ghandi et al. 2014b), and the score for each sequence is the sum of the weights for all gapped k-mers in a sequence. These weights quantify the contribution of each gapped k-mer to chromatin accessibility. To map these gapped k-mer weights to interpretable TF activity, after training gkm-SVM models, we extracted TF models and their inferred activity using gkm-PWM, which infers the frequency of TFBS position weight matrices (PWMs) by minimizing the error between the observed and generated weight vectors. All of the 10 STAD ATAC samples with more than 10,000 distal peaks and AUROC > 0.9 detect some activation of RUNX or AP-1 when trained against normal stomach DNase-seq (ENCODE: ENCSR782SSS): Seven detect AP-1 (TCGA.CD.A48C, TCGA.VQ.A94O, TCGA.VQ.A8PJ, TCGA.BR.A4J6, TCGA.BR.A4CS, TCGA.HF.A5NB, TCGA.BR.A4J4), and seven detect RUNX (TCGA.VQ.A94O, TCGA.VQ.A91W, TCGA.BR.A4J6, TCGA.BR.A4IY, TCGA.HF.A5NB, TCGA.BR.A4J4, TCGA.CD.A486) (Supplemental Fig. S5). For the GC cell lines, to generate a more compact set of TFBS motifs, we trained each set of positive ATAC distal peaks versus each other (300 pairs of experiments, n = 2000 differentially active sequences in each of the two cell types, median AUROC = 0.922) (Supplemental Fig. S27) and used the 19 most commonly detected motifs as PWMs to extract TF activity (AP-1, RUNX, AP2, NFKB, ZEB (AREB6), NFI, ONECUT, EBOX, EHF, TCF/LEF, TEAD, GRHL, HNF4A, HNF1B, HOXB13, FOX, KLF, SOX, GATA) (Fig. 1E; Supplemental Fig. S28). Although many of these motifs are detected in other cell types (McClymont et al. 2018), the combinations are specific to endodermal lineages (Luo et al. 2023). Clustering in the inferred motif activity space generates a PCA plot with the same grouping detected in the ATAC-seq signal (Fig. 1A).
ATAC-seq analysis around DE genes
We calculated the ATAC-seq signal in peaks within 50,000 bp around the TSS of DE genes in Epi and Mes (higher in Epi, higher in Mes). The average activity in flanking peaks averaged over all Epi, Mes, and Intermediate cell lines is shown in Figure 6A. Similar results were found using peaks within 100,000, 150,000, 200,000, or 250,000 bp from the TSS.
ChIP-seq data
ChIP-seq samples from ENCODE were used in the genome browser tracks: ZEB1 (ENCODE ENCSR000BVN) and SMAD4 (ENCODE ENCSR826YMT). ChIP-seq data for RUNX2 and JUN in Figure 1F are from Bevill et al. (2023). ChIP-seq data for KLF5 in AGS are from Liu et al. (2020), and ChIP-seq data for GATA4 and GATA6 in AGS are from Chia et al. (2015) with raw data available under GEO accession number GSE51705.
RNA-seq data processing
GC cell line RNA-seq
The raw GC cell line RNA-seq data analyzed in this study were previously reported (Xing et al. 2020; Sheng et al. 2021; Ho et al. 2023; Xu et al. 2023) and uploaded to GEO under accession numbers GSE266159, GSE157750, and GSE85465. To normalize FPKM values, we divided them by the sample's upper-quartile (75th percentile) expression value and multiplied them by the average upper-quartile values across all samples to scale (upper-quartile normalization), and then, they were log2-transformed.
TCGA-STAD and TCGA-normal RNA-seq
TCGA-STAD (stomach adenocarcinoma) and TCGA-Normal (normal adjacent stomach tissue) RNA-seq HTSeq counts were downloaded from the TCGA GDC portal (https://portal.gdc.cancer.gov/) (Anders et al. 2015). Three hundred fifty-six tumor (TCGA-STAD) and 32 normal stomach (TCGA-Normal) samples were initially obtained. TCGA-STAD and TCGA-Normal samples with a higher correlation to GTEx esophagus than to GTEx stomach tissue RNA-seq signature were removed (with a method similar to the GTEx correlation analysis described below). The remaining 322 TCGA-STAD and 17 TCGA-Normal samples were upper-quartile-normalized over the set of protein-coding genes as labeled by GENCODE V35 annotation (Frankish et al. 2019).
Chromatin accessibility analysis of Mes-like GC and normal ENCODE fibroblast and stomach
To compare chromatin accessibility profiles, we generated a union set of all distal peaks of ENCODE stomach (n = 12) and primary fibroblast (n = 30), as well as 25 GC cell lines to perform PCA. We trained gkm-SVM models on the 2000 most differentially accessible peaks in all pairs of five Mes-like GC cell lines and 30 ENCODE primary fibroblasts and ranked nonsimilar motifs by their average gkm-PWM Z-score.
RNA-seq analysis
Tissue-specific protein-coding genes
To reduce noise, RNA-seq analyses were performed using 11,312 “tissue-specific” genes, which are derived from GTEx RNA-seq profiles of 54 healthy human tissues (GTEx portal: GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_median_tpm.gct.gz). To define tissue-specific genes, we calculated the number of GTEx tissues, in which a gene has a TPM expression above the median expression in that tissue (min = 0, max = 54). If this number is zero, the gene is lowly expressed in all tissues, and if this number is 54, the gene is ubiquitously expressed (e.g., ribosomal genes). Tissue-specific genes are then defined as the subset of protein-coding genes that are neither lowly expressed nor ubiquitously expressed in all GTEx healthy human tissues. Protein-coding genes are found according to GENCODE V35 annotation.
PCA of RNA-seq data
PCA was performed using prcomp() and drawn with ggplot2 in R (R Core Team 2023). For the joint cell line and primary tissue PCA, GC cell line and TCGA-STAD FPKM expression values were upper-quartile-normalized together over the set of tissue-specific genes. Genes having the highest (positive or negative) weights in the PCA were obtained using R prcomp$rotation[, 1] and prcomp$rotation[, 2] for PC1 and PC2, in which the top 100 genes were used for gene set analysis. We used upper-quartile-normalized RNA-seq from LPS141 (Chen et al. 2019) to add this cell line to the PCA in Supplemental Figure S6, which shows that the transcriptional state of LPS141 is very similar to the Mes-like GC cell lines.
DE genes in Mes-like versus Epi-like cell lines
DE genes between the Mes-like and Epi-like GC cell lines were found using the edgeR (Robinson et al. 2010) package in R. The list of genes was limited to 11,312 tissue-specific protein-coding genes, as described above. One thousand seven hundred three (including 153 TFs) were found to be upregulated in Mes-like GC cell lines (|log2FC| > 2, FDR < 0.05) (Supplemental Table S1), whereas 976 genes (including 58 TFs) have a higher expression in Epi-like cell lines (Supplemental Table S2). The list of all 1639 human TFs was retrieved from http://humantfs.ccbr.utoronto.ca/download.php (version 1.01) (Lambert et al. 2018).
Survival analysis in ACRG and TCGA-STAD
Gene expression values for 300 ACRG GC patients were downloaded from GEO accession number GSE62254. Patients were stratified into Mes-like (n = 150) and Epi-like (n = 150) groups based on the median difference in the mean expression values of Mes and Epi DE TFs. Similarly, TCGA-STAD patients (n = 315) were split into two groups using the median expression difference value. Plots and P-values were produced using the survminer (https://github.com/kassambara/survminer), ggsurvfit (https://github.com/pharmaverse/ggsurvfit), and survival (https://github.com/therneau/survival) R packages.
Gene set analysis
GSEA software (Mootha et al. 2003; Subramanian et al. 2005) with hallmark gene sets (Liberzon et al. 2015) was used to identify the biological function of DE genes in Mes-like versus Epi-like GC (default GSEA parameters). Additionally, MSigDB (Liberzon et al. 2011) gene sets H (hallmark), C2 (curated gene sets), C5 (ontology gene sets), and C6 (oncogenic gene sets) were used with the top 100 PC$rotation (see section PCA of RNA-seq in above) to find enriched gene sets. Hypergeometric test calculated the P-values, which were then adjusted using p.adjust(method = “fdr”) in R.
DE genes in Mes-like GC versus ENCODE fibroblast
RNA-seq data for normal human fibroblast were downloaded from the ENCODE portal and were upper-quartile-normalized with the GC cell line RNA-seq profiles. Normalization and the DE gene analysis were performed similar to what described above (FDR < 0.01, log2FC > 2), in which 1133 and 545 genes are upregulated in Mes-like GC and ENCODE fibroblast samples, respectively. One hundred eleven of the upregulated genes in the Mes-like group and 37 in the ENCODE fibroblast group are TFs. To compare the expression of these DE TFs in normal stomach and normal fibroblast, we used GTEx normal stomach and GTEx normal fibroblast to rank the expression values of these DE TFs (between Mes and ENCODE-fibroblast) in the independent GTEx stomach and fibroblast expression profiles.
CN and mutation analyses
TCGA-STAD mutation rates were downloaded from cBioPortal (Cerami et al. 2012; Gao et al. 2013; https://www.cbioportal.org/study/summary?id=stad_tcga_pan_can_atlas_2018). Of the TCGA-STAD samples for which the RNA-seq data were available, 255 samples are microsatellite stable (MSS) and were used for further analysis, whereas the MSI samples were removed because of the possibility of having more passenger mutations. The genes selected in the plot are TFs DE between the Mes-like and Epi-like GC cell lines. The x-axis is the log2 fold-change of FPKM-UQ values between the 14 samples with a CDH1 nonsynonymous or splice-site mutation and the 241 samples without; the y-axis P-values were calculated using the t-test.
CN alteration rates for all human TFs in the TCGA-STAD (Pan-Cancer) samples were downloaded from cBioPortal and include CN deletion and CN amplification events as calculated by GISTIC (Mermel et al. 2011). GISTIC assigns an integer “score” between −2 and +2 to each gene depending on the number of DNA fragments aligned to it in the sequencing data. GISTIC “scores” “−2” and “−1” represent deep and shallow DNA deletions (possibly homozygous and heterozygous deletions), whereas “+2” and “+1” indicate deep and shallow DNA amplifications, respectively. A GISTIC “score” of “0” for a gene in a sample means that the gene DNA CN is not changed and is normal (i.e., the CN is two in a healthy diploid human genome).
Whole-genome sequencing (WGS) data of GC cell lines were processed as previously described (Xing et al. 2020). Briefly, CN variations were identified using CNVkit with default parameters (bcbio-nextgen v0.9.3) (Talevich et al. 2016). As GC cell lines have no matched germline samples, CNVs were call against a nonmatched normal sample. We filtered high-confidence CN predictions (n = 2061) by removing protein-coding genes whose expression correlation with CN across cell lines is less than 0.5. We calculated CN differences between the 14 Epi-like GC cell lines and the combined six Mes-like and five Intermediate cell lines, and we plotted the average CN differences for each gene in the two groups, that is, mean(NonEpi CN) − mean(Epi CN). The y-axis is the P-value for the difference between Epi and NonEpi CN scores using the Mann–Whitney U test. We also analyzed SNVs in the GC cell lines with SmuRF (Huang et al. 2020) and found mutations in GATA6 and ZFPM2 (also known as friend of GATA family 2 [FOG2]) in 4/6 of the Mes-like cell lines, but their frequency was close to the threshold for common SNPs, and after filtering, the P-values were not significant.
To compare the nonsynonymous mutation burden across groups of GC cell lines, we performed variant calling on the WGS of GC cell lines using SMuRF (Huang et al. 2020). As no matched germline is available for these cell lines, we filtered out common population variants from the dbSNP database (AF > 1%). Then, we calculated the number of protein-altering mutations (missense, truncating, start and stop codon gain/loss, and splice region variants) in each cell line (Supplemental Table S5).
To calculate the CN burden in GC cell lines, we converted the CNVkit output to a GISTIC-like integer score (ranging from −2 to +2; described above). For each cell line, the absolute values of gene CN scores are averaged to report the cell line's overall CN status.
RNA-seq and ATAC-seq correlations
Correlation analysis using GTEx tissue RNA-seq data
GTEx RNA-seq TPM values for 54 healthy human tissues were retrieved as described above. To compare GC cell line RNA-seq with GTEx, the cell line's FPKM values were converted to TPM. To preserve space in the figures, the TPM expression values for all GTEx brain tissues were aggregated (averaged). The mean expression for each tissue-specific gene (described above) for the three cell line groups (Mes, Epi, Intermediate) was calculated, and this resulted in a 11,312 × 3 TPM expression matrix for the cell lines. Then the Pearson's correlation was calculated between the GC cell line groups and GTEx tissues, using TPM expression values.
Correlation heatmap with ENCODE fibroblast and stomach RNA-seq data
The following fibroblast and stomach RNA-seq profiles were downloaded from the ENCODE portal: HT1080 (ENCFF754UAP), bronchus fibroblast of lung (ENCFF716LRF), fibroblast skin of abdomen (ENCFF010QUB), fibroblast skin of scalp (ENCFF385POO), stomach 3-yr-old child (ENCFF299YCQ), stomach 37-yr-old adult (ENCFF683JSC), and stomach 34-yr-old adult (ENCFF547FBP). Upper-quartile-normalized FPKM values of ENCODE samples, GC cell lines, and TCGA-STAD were then compared in a heatmap (Pearson's correlation). The 11,312 tissue-specific genes (described above) were used to calculate the correlation. In the scatter plot, correlation values for each individual sample are shown.
Correlation analysis with ENCODE fibroblast and stomach DNase-seq data
The following fibroblast and stomach DNase-seq profiles were downloaded from the ENCODE portal and processed as described above: cardiac fibroblast (ENCSR000ENI), lung fibroblast (ENCSR000EPR), ELR fibroblast cell line (ENCSR240TPI), HT1080 fibrosarcoma cell line (ENCSR000FDI), stomach 34-yr-old adult (ENCSR782SSS), stomach 54-yr-old adult (ENCSR163PKT), and stomach 3-yr-old child (ENCSR246PXX). gkm-SVM models were trained on ENCODE DNase-seq, GC cell line ATAC-seq, and TCGA-STAD ATAC-seq profiles, as described above. The gkm-SVM output for each sample is a weight vector corresponding to 411/2 k-mers (A/T/C/G 11-mers), where the weight value and its sign indicate how overrepresented or underrepresented the k-mer (TF binding site) is in the chromatin accessibility data (ATAC or DNase-seq). To find the similarities between the chromatin accessibility profiles, a heatmap (Pearson's correlation) was drawn using the ENCODE, cell line, and TCGA-STAD samples. To calculate the correlation for each group (i.e., Mes-like, Epi-like, TCGA-STAD), their mean gkm-SVM weight vectors were used for each group of samples. In the scatter plot, correlation values for each individual sample are shown. In Figure 4, D and E, lung fibroblast (ENCSR000EPR) and adult stomach (ENCSR782SSS) were used.
scRNA-seq data processing and analysis
scRNA-seq raw counts for 24 patients (gastric tumor and normal adjacent tissue) were downloaded from the Kim et al. (2022) study under GEO accession number GSE150290. Cells having (1) fewer than 500 detected transcripts, (2) more than 20,000 transcripts, (3) fewer than 500 detected genes, or (4) >10% mitochondrial or hemoglobin genes were filtered out for being low quality. This resulted in an expression profile of a total of about 117,000 cells derived from tumor and normal adjacent tissue of 24 GC patients. Log-normalization was performed using Seurat (Hao et al. 2021), and UMAP was used for dimensionality reduction. Clusters were defined by running a Gaussian mixture model (GMM) over the UMAP space using the R uwot package (n_components = 2, n_neighbors = 30) (McInnes et al. 2020). To find the correlation and similarities between scRNA-seq populations (clusters) and GC cell line groups, the correlation of scRNA and bulk RNA was calculated for each cell. The average RNA-seq expression for the Epi-like or Mes-like groups of cell lines over their DE genes (2679 genes) was calculated. Then for each cell, the Pearson's correlation of the scRNA-seq expression values (for 2679 DE genes) and the average bulk RNA-seq expression of Mes-like and Epi-like cell lines was calculated.
Kim et al. (2022) discovered several cell populations, including immune cells, fibroblasts, and various gastric cell types. We saw a similar pattern of marker genes in our analysis, in which clusters 1, 3, and 4 express immune-related markers such as CD74, CD83, PTPRC (previously known as CD45), IL32, and various MHC/HLA class II variants. Clusters 2 and 5 express gastrointestinal gene markers such as TFF1, TFF2, TFF3, PGC, and REG4, similar to what was discovered by Kim et al. (2022), whereas cluster 6 expresses fibrotic markers, including COL1A1, COL1A2, COL3A1, COL6A1, COL6A2, PDGFRA, FN1, and MMP2. This indicates that single-cell clusters 2 and 5 include stomach cells, whereas cluster 6 is composed of fibroblasts. To define a more systematic list of highly expressed gene markers for fibroblast (or stomach) tissue, we used the same GTEx 54 healthy tissue RNA-seq profiles as described above. We sorted the TPM expression values of 11,312 tissue-specific genes (described above) in GTEx fibroblast (or stomach). We used the top 100 highly expressed genes as the gene markers for fibroblast (or stomach). To find the biological interpretation of each single-cell cluster, we used these gene markers. For every single cell, the average expression over the top 100 genes (fibroblast or stomach) was calculated, and the boxplot (Fig. 5D,E) was drawn based on the single-cell clusters, where clusters 2 and 5 were combined as they were both similarly correlated with the same group of GC cell lines. The average expression values across each cluster (in the boxplots) are compared using a t-test, where P < 10−8. Thus, gene expression in Epi-like cell lines is highly correlated to that of stomach cells, unlike Mes-like GC cell lines, which have a high correlation with the fibroblast single-cell population.
LE model predictions
CTCF loop predictions shown in Figures 3C, 6D, and 6E use LE model predictions (Xi and Beer 2021) using CTCF ChIP-seq in endoderm (Luo et al. 2023). The LE model uses orientation and binding strength information from ChIP-seq to predict loop probability, is consistent with CTCF ChIA-PET loop measurements, and generalizes better than more complicated CTCF loop prediction methods (Xi and Beer 2018).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE264550 and GSE266159. Source files for differential accessibility and expression analysis are included as Supplemental Code, and both code and gkm-SVM models are available at http://beerlab.org/gccl.
Competing interest statement
P.T. has stock and other ownership interests in Tempus Healthcare, previous research funding from Kyowa Hakko Kirin and Thermo Fisher Scientific, and patents/other intellectual property through the Agency for Science and Technology Research, Singapore (all outside the submitted work). All remaining authors declare no competing interests.
Acknowledgments
This work was supported by the following grants: M.R.-M., D.S., and M.A.B. were supported by National Institutes of Health–National Human Genome Research Institute grant R01 HG012367 to M.A.B. W.H., Y.A.G., S.W.T.H., A.J.S., and P.T. are supported by National Medical Research Council grant MOH-000967-00 (MOH-STaR21jun-0001) and by the National Research Foundation, Singapore, and Singapore Ministry of Health's National Medical Research Council under its Open Fund-Large Collaborative grant (“OF-LCG”; MOH-OFLCG18May-0003). The cell line GES1 was a gift from Alfred Cheng from the Chinese University of Hong Kong. We thank Salvador Casaní-Galdón and Brad Bernstein for providing the ChIP-seq bigwigs from Bevill et al. (2023). We thank Roger Sik Yin Foo and Chukwuemeka George Anene-Nzelu for assisting with ATAC-seq library preparation and sequencing.
Author contributions: Most analyses were performed and designed by M.R.-M. and M.A.B., with contributions from A.J.S., W.H., Y.A.G., and D.S. Overall study design was conceived by M.A.B. and A.J.S. All authors provided editorial and intellectual input.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278565.123.
References
- ↵Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn C-O, Heidecke C-D, Friess H, Büchler M, Evert M, Lerch MM, 2012. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 61: 439–448. 10.1136/gutjnl-2011-300060
- ↵Ajani JA, Lee J, Sano T, Janjigian YY, Fan D, Song S. 2017. Gastric adenocarcinoma. Nat Rev Dis Primer 3: 17036. 10.1038/nrdp.2017.36
- ↵Amanchy R, Kandasamy K, Mathivanan S, Periaswamy B, Reddy R, Yoon W-H, Joore J, Beer MA, Cope L, Pandey A. 2011. Identification of novel phosphorylation motifs through an integrative computational and experimental analysis of the human phosphoproteome. J Proteomics Bioinform 4: 22–35. 10.4172/jpb.1000163
- ↵Anders S, Pyl PT, Huber W. 2015. HTSeq: a python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. 10.1093/bioinformatics/btu638
- ↵Aran D, Sirota M, Butte AJ. 2015. Systematic pan-cancer analysis of tumour purity. Nat Commun 6: 8971. 10.1038/ncomms9971
- ↵Badgwell B, Das P, Ajani J. 2017. Treatment of localized gastric and gastroesophageal adenocarcinoma: the role of accurate staging and preoperative therapy. J Hematol Oncol 10: 149. 10.1186/s13045-017-0517-9
- ↵Bakiri L, Macho-Maschler S, Custic I, Niemiec J, Guío-Carrión A, Hasenfuss SC, Eger A, Müller M, Beug H, Wagner EF. 2015. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ 22: 336–350. 10.1038/cdd.2014.157
- ↵Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard B, Hinoue T, Laird PW, Curtis C, Shen H, 2014. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513: 202–209. 10.1038/nature13480
- ↵Beer MA. 2017. Predicting enhancer activity and variant impact using gkm-SVM. Hum Mutat 38: 1251–1258. 10.1002/humu.23185
- ↵Beer MA, Shigaki D, Huangfu D. 2020. Enhancer predictions and genome-wide regulatory circuits. Annu Rev Genomics Hum Genet 21: 37–54. 10.1146/annurev-genom-121719-010946
- ↵Berlth F, Bollschweiler E, Drebber U, Hoelscher AH, Moenig S. 2014. Pathohistological classification systems in gastric cancer: diagnostic relevance and prognostic value. World J Gastroenterol 20: 5679–5684. 10.3748/wjg.v20.i19.5679
- ↵Bevill SM, Casaní-Galdón S, El Farran CA, Cytrynbaum EG, Macias KA, Oldeman SE, Oliveira KJ, Moore MM, Hegazi E, Adriaens C, 2023. Impact of supraphysiologic MDM2 expression on chromatin networks and therapeutic responses in sarcoma. Cell Genomics 3: 100321. 10.1016/j.xgen.2023.100321
- ↵Bordignon P, Bottoni G, Xu X, Popescu AS, Truan Z, Guenova E, Kofler L, Jafari P, Ostano P, Röcken M, 2019. Dualism of FGF and TGF-β signaling in heterogeneous cancer-associated fibroblast activation with ETV1 as a critical determinant. Cell Rep 28: 2358–2372.e6. 10.1016/j.celrep.2019.07.092
- ↵Brabletz T, Kalluri R, Nieto MA, Weinberg RA. 2018. EMT in cancer. Nat Rev Cancer 18: 128–134. 10.1038/nrc.2017.118
- ↵Byles V, Zhu L, Lovaas JD, Chmilewski LK, Wang J, Faller DV, Dai Y. 2012. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 31: 4619–4629. 10.1038/onc.2011.612
- ↵Cai Y, Sablina AA. 2016. Cancer-associated chromosomal deletions: size makes a difference. Cell Cycle 15: 2850–2851. 10.1080/15548627.2016.1204869
- ↵Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, 2012. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. 10.1158/2159-8290.CD-12-0095
- ↵Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adélaïde J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, 2006. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 25: 2273–2284. 10.1038/sj.onc.1209254
- ↵Chen Y, Xu L, Mayakonda A, Huang M-L, Kanojia D, Tan TZ, Dakle P, Lin RY-T, Ke X-Y, Said JW, 2019. Bromodomain and extraterminal proteins foster the core transcriptional regulatory programs and confer vulnerability in liposarcoma. Nat Commun 10: 1353. 10.1038/s41467-019-09257-z
- ↵Chia N-Y, Deng N, Das K, Huang D, Hu L, Zhu Y, Lim KH, Lee M-H, Wu J, Sam XX, 2015. Regulatory crosstalk between lineage-survival oncogenes KLF5, GATA4 and GATA6 cooperatively promotes gastric cancer development. Gut 64: 707–719. 10.1136/gutjnl-2013-306596
- ↵Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, 2018. The chromatin accessibility landscape of primary human cancers. Science 362: eaav1898. 10.1126/science.aav1898
- ↵Cristescu R, Lee J, Nebozhyn M, Kim K-M, Ting JC, Wong SS, Liu J, Yue YG, Wang J, Yu K, 2015. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 21: 449–456. 10.1038/nm.3850
- ↵The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247
- ↵Fang F, Chang R, Yu L, Lei X, Xiao S, Yang H, Yang L-Y. 2015. MicroRNA-188-5p suppresses tumor cell proliferation and metastasis by directly targeting FGF5 in hepatocellular carcinoma. J Hepatol 63: 874–885. 10.1016/j.jhep.2015.05.008
- ↵Fodde R. 2002. The APC gene in colorectal cancer. Eur J Cancer 38: 867–871. 10.1016/S0959-8049(02)00040-0
- ↵Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, 2019. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47: D766–D773. 10.1093/nar/gky955
- ↵Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1–pl1. 10.1126/scisignal.2004088
- ↵Gate RE, Cheng CS, Aiden AP, Siba A, Tabaka M, Lituiev D, Machol I, Gordon MG, Subramaniam M, Shamim M, 2018. Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat Genet 50: 1140–1150. 10.1038/s41588-018-0156-2
- ↵Ghandi M, Lee D, Mohammad-Noori M, Beer MA. 2014a. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol 10: e1003711. 10.1371/journal.pcbi.1003711
- ↵Ghandi M, Mohammad-Noori M, Beer MA. 2014b. Robust k-mer frequency estimation using gapped k-mers. J Math Biol 69: 469–500. 10.1007/s00285-013-0705-3
- ↵Ghandi M, Mohammad-Noori M, Ghareghani N, Lee D, Garraway L, Beer MA. 2016. gkmSVM: an R package for gapped-kmer SVM. Bioinformatics 32: 2205–2207. 10.1093/bioinformatics/btw203
- ↵Guneri-Sozeri PY, Özden-Yılmaz G, Kisim A, Cakiroglu E, Eray A, Uzuner H, Karakülah G, Pesen-Okvur D, Senturk S, Erkek-Ozhan S. 2023. FLI1 and FRA1 transcription factors drive the transcriptional regulatory networks characterizing muscle invasive bladder cancer. Commun Biol 6: 199. 10.1038/s42003-023-04561-3
- ↵Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, 2021. Integrated analysis of multimodal single-cell data. Cell 184: 3573–3587.e29. 10.1016/j.cell.2021.04.048
- ↵Ho SWT, Sheng T, Xing M, Ooi WF, Xu C, Sundar R, Huang KK, Li Z, Kumar V, Ramnarayanan K, 2023. Regulatory enhancer profiling of mesenchymal-type gastric cancer reveals subtype-specific epigenomic landscapes and targetable vulnerabilities. Gut 72: 226–241. 10.1136/gutjnl-2021-326483
- ↵Huang W, Guo YA, Chang MM, Skanderup AJ. 2020. Ensemble-based somatic mutation calling in cancer genomes. In Bioinformatics for cancer immunotherapy: methods and protocols (ed. Boegel S), pp. 37–46. Springer, New York. 10.1007/978-1-0716-0327-7_3
- ↵Kalluri R, Zeisberg M. 2006. Fibroblasts in cancer. Nat Rev Cancer 6: 392–401. 10.1038/nrc1877
- ↵Karin M, Liu Z, Zandi E. 1997. AP-1 function and regulation. Curr Opin Cell Biol 9: 240–246. 10.1016/S0955-0674(97)80068-3
- ↵Kim J, Park C, Kim KH, Kim EH, Kim H, Woo JK, Seong JK, Nam KT, Lee YC, Cho SY. 2022. Single-cell analysis of gastric pre-cancerous and cancer lesions reveals cell lineage diversity and intratumoral heterogeneity. NPJ Precis Oncol 6: 9. 10.1038/s41698-022-00251-1
- ↵Kreimer A, Zeng H, Edwards MD, Guo Y, Tian K, Shin S, Welch R, Wainberg M, Mohan R, Sinnott-Armstrong NA, 2017. Predicting gene expression in massively parallel reporter assays: a comparative study. Hum Mutat 38: 1240–1250. 10.1002/humu.23197
- ↵Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT. 2018. The human transcription factors. Cell 172: 650–665. 10.1016/j.cell.2018.01.029
- ↵Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923
- ↵Lee D. 2016. LS-GKM: a new gkm-SVM for large-scale datasets. Bioinformatics 32: 2196–2198. 10.1093/bioinformatics/btw142
- ↵Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, Beer MA. 2015. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet 47: 955–961. 10.1038/ng.3331
- ↵Lei Z, Tan IB, Das K, Deng N, Zouridis H, Pattison S, Chua C, Feng Z, Guan YK, Ooi CH, 2013. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 145: 554–565. 10.1053/j.gastro.2013.05.010
- ↵Li QV, Dixon G, Verma N, Rosen BP, Gordillo M, Luo R, Xu C, Wang Q, Soh C-L, Yang D, 2019. Genome-scale screens identify JNK–JUN signaling as a barrier for pluripotency exit and endoderm differentiation. Nat Genet 51: 999–1010. 10.1038/s41588-019-0408-9
- ↵Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. 2011. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740. 10.1093/bioinformatics/btr260
- ↵Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. 2015. The molecular signatures database hallmark gene set collection. Cell Syst 1: 417–425. 10.1016/j.cels.2015.12.004
- ↵Liu Y, Guo B, Aguilera-Jimenez E, Chu VS, Zhou J, Wu Z, Francis JM, Yang X, Choi PS, Bailey SD, 2020. Chromatin looping shapes KLF5-dependent transcriptional programs in human epithelial cancers. Cancer Res 80: 5464–5477. 10.1158/0008-5472.CAN-20-1287
- ↵Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653
- ↵Lovisa S. 2021. Epithelial-to-mesenchymal transition in fibrosis: concepts and targeting strategies. Front Pharmacol 12: 737570. 10.3389/fphar.2021.737570
- ↵Lui FH, Tuan B, Swenson SL, Wong RJ. 2014. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992–2009 SEER data. Dig Dis Sci 59: 3027–3034. 10.1007/s10620-014-3275-3
- ↵Luo W, Fedda F, Lynch P, Tan D. 2018. CDH1 gene and hereditary diffuse gastric cancer syndrome: molecular and histological alterations and implications for diagnosis and treatment. Front Pharmacol 9: 1421. 10.3389/fphar.2018.01421
- ↵Luo J, Chen X-Q, Li P. 2019. The role of TGF-β and its receptors in gastrointestinal cancers. Transl Oncol 12: 475–484. 10.1016/j.tranon.2018.11.010
- ↵Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, Myers Z, Sud P, Jou J, Lin K, 2020. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res 48: D882–D889. 10.1093/nar/gkz1062
- ↵Luo R, Yan J, Oh JW, Xi W, Shigaki D, Wong W, Cho HS, Murphy D, Cutler R, Rosen BP, 2023. Dynamic network-guided CRISPRi screen identifies CTCF-loop-constrained nonlinear enhancer gene regulatory activity during cell state transitions. Nat Genet 55: 1336–1346. 10.1038/s41588-023-01450-7
- ↵Martinelli P, Pau ECS, Cox T, Sainz B, Dusetti N, Greenhalf W, Rinaldi L, Costello E, Ghaneh P, Malats N, 2017. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 66: 1665–1676. 10.1136/gutjnl-2015-311256
- ↵Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, Mularoni L, Pich O, Bonet J, Kranas H, 2020. A compendium of mutational cancer driver genes. Nat Rev Cancer 20: 555–572. 10.1038/s41568-020-0290-x
- ↵McClymont SA, Hook PW, Soto AI, Reed X, Law WD, Kerans SJ, Waite EL, Briceno NJ, Thole JF, Heckman MG, 2018. Parkinson-associated SNCA enhancer variants revealed by open chromatin in mouse dopamine neurons. Am J Hum Genet 103: 874–892. 10.1016/j.ajhg.2018.10.018
- ↵McInnes L, Healy J, Melville J. 2020. UMAP: Uniform Manifold Approximation and Projection for dimension reduction. arXiv: 1802.03426 [stat.ML]. http://arxiv.org/abs/1802.03426
- ↵Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. 2011. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12: R41. 10.1186/gb-2011-12-4-r41
- ↵Mo A, Luo C, Davis FP, Mukamel EA, Henry GL, Nery JR, Urich MA, Picard S, Lister R, Eddy SR, 2016. Epigenomic landscapes of retinal rods and cones. eLife 5: e11613. 10.7554/eLife.11613
- ↵Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, 2003. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273. 10.1038/ng1180
- ↵Nissen NI, Karsdal M, Willumsen N. 2019. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J Exp Clin Cancer Res 38: 115. 10.1186/s13046-019-1110-6
- ↵Niu D-F, Kondo T, Nakazawa T, Oishi N, Kawasaki T, Mochizuki K, Yamane T, Katoh R. 2012. Transcription factor Runx2 is a regulator of epithelial–mesenchymal transition and invasion in thyroid carcinomas. Lab Invest 92: 1181–1190. 10.1038/labinvest.2012.84
- ↵Oh SC, Sohn BH, Cheong J-H, Kim S-B, Lee JE, Park KC, Lee SH, Park J-L, Park Y-Y, Lee H-S, 2018. Clinical and genomic landscape of gastric cancer with a mesenchymal phenotype. Nat Commun 9: 1777. 10.1038/s41467-018-04179-8
- ↵Park S, Lee J, Kim YH, Park J, Shin J-W, Nam S. 2016. Clinical relevance and molecular phenotypes in gastric cancer, of TP53 mutations and gene expressions, in combination with other gene mutations. Sci Rep 6: 34822. 10.1038/srep34822
- ↵Piersma B, Hayward M-K, Weaver VM. 2020. Fibrosis and cancer: a strained relationship. Biochim Biophys Acta Rev Cancer 1873: 188356. 10.1016/j.bbcan.2020.188356
- ↵R Core Team. 2023. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna https://www.R-project.org/.
- ↵Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. 10.1038/nature14248
- ↵Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. 10.1093/bioinformatics/btp616
- ↵Roche J. 2018. The epithelial-to-mesenchymal transition in cancer. Cancers (Basel) 10: 52. 10.3390/cancers10020052
- ↵Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, 2020. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20: 174–186. 10.1038/s41568-019-0238-1
- ↵Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, Walter M, Fehm T, Solomayer E, Riess O, Wallwiener D, 2006. Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer Res 66: 5278–5286. 10.1158/0008-5472.CAN-05-4610
- ↵Sheng T, Ho SWT, Ooi WF, Xu C, Xing M, Padmanabhan N, Huang KK, Ma L, Ray M, Guo YA, 2021. Integrative epigenomic and high-throughput functional enhancer profiling reveals determinants of enhancer heterogeneity in gastric cancer. Genome Med 13: 158. 10.1186/s13073-021-00970-3
- ↵Shigaki D, Adato O, Adhikar AN, Dong S, Hawkins-Hooker A, Inoue F, Juven-Gershon T, Kenlay H, Martin B, Patra A, 2019. Integration of multiple epigenomic marks improves prediction of variant impact in saturation mutagenesis reporter assay. Hum Mutat 40: 1280–1291. 10.1002/humu.23797
- ↵Shin JY, Beckett JD, Bagirzadeh R, Creamer TJ, Shah AA, McMahan Z, Paik JJ, Sampedro MM, MacFarlane EG, Beer MA, 2019. Epigenetic activation and memory at a TGFB2 enhancer in systemic sclerosis. Sci Transl Med 11: eaaw0790. 10.1126/scitranslmed.aaw0790
- ↵Sohn BH, Hwang J-E, Jang H-J, Lee H-S, Oh SC, Shim J-J, Lee K-W, Kim EH, Yim SY, Lee SH, 2017. Clinical significance of four molecular subtypes of gastric cancer identified by the Cancer Genome Atlas Project. Clin Cancer Res 23: 4441–4449. 10.1158/1078-0432.CCR-16-2211
- ↵Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. 10.1073/pnas.0506580102
- ↵Talevich E, Shain AH, Botton T, Bastian BC. 2016. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 12: e1004873. 10.1371/journal.pcbi.1004873
- ↵Tan IB, Ivanova T, Lim KH, Ong CW, Deng N, Lee J, Tan SH, Wu J, Lee MH, Ooi CH, 2011. Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology 141: 476–485.e11. 10.1053/j.gastro.2011.04.042
- ↵Taylor VM, Ko LK, Hwang JH, Sin M-K, Inadomi JM. 2015. Gastric cancer in Asian American populations: a neglected health disparity. Asian Pac J Cancer Prev 15: 10565–10571. 10.7314/APJCP.2014.15.24.10565
- ↵Vecchi M, Nuciforo P, Romagnoli S, Confalonieri S, Pellegrini C, Serio G, Quarto M, Capra M, Roviaro GC, Contessini Avesani E, 2007. Gene expression analysis of early and advanced gastric cancers. Oncogene 26: 4284–4294. 10.1038/sj.onc.1210208
- ↵Verrecchia F, Chu M-L, Mauviel A. 2001. Identification of novel TGF-β/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem 276: 17058–17062. 10.1074/jbc.M100754200
- ↵Wang X, Xu J, Zhang B, Hou Y, Song F, Lyu H, Yue F. 2021. Genome-wide detection of enhancer-hijacking events from chromatin interaction data in rearranged genomes. Nat Methods 18: 661–668. 10.1038/s41592-021-01164-w
- ↵Xi W, Beer MA. 2018. Local epigenomic state cannot discriminate interacting and non-interacting enhancer–promoter pairs with high accuracy. PLoS Comput Biol 14: e1006625. 10.1371/journal.pcbi.1006625
- ↵Xi W, Beer MA. 2021. Loop competition and extrusion model predicts CTCF interaction specificity. Nat Commun 12: 1046. 10.1038/s41467-021-21368-0
- ↵Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, Ni Z, Zhang B, Zhang D, Luo F, 2020. FGF/FGFR signaling in health and disease. Signal Transduct Target Ther 5: 181. 10.1038/s41392-020-00222-7
- ↵Xing M, Ooi WF, Tan J, Qamra A, Lee P-H, Li Z, Xu C, Padmanabhan N, Lim JQ, Guo YA, 2020. Genomic and epigenomic EBF1 alterations modulate TERT expression in gastric cancer. J Clin Invest 130: 3005–3020. 10.1172/JCI126726
- ↵Xu C, Huang KK, Law JH, Chua JS, Sheng T, Flores NM, Pizzi MP, Okabe A, Tan ALK, Zhu F, 2023. Comprehensive molecular phenotyping of ARID1A-deficient gastric cancer reveals pervasive epigenomic reprogramming and therapeutic opportunities. Gut 72: 1651–1663. 10.1136/gutjnl-2022-328332
- ↵Yan J, Qiu Y, Ribeiro dos Santos AM, Yin Y, Li YE, Vinckier N, Nariai N, Benaglio P, Raman A, Li X, 2021. Systematic analysis of binding of transcription factors to noncoding variants. Nature 591: 147–151. 10.1038/s41586-021-03211-0
- ↵Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, Campbell K, Cano A, Casanova J, Christofori G, 2020. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat Rev Mol Cell Biol 21: 341–352. 10.1038/s41580-020-0237-9
- ↵Yu K, Chen B, Aran D, Charalel J, Yau C, Wolf DM, van ‘t Veer LJ, Butte AJ, Goldstein T, Sirota M. 2019. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat Commun 10: 3574. 10.1038/s41467-019-11415-2
- ↵Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137. 10.1186/gb-2008-9-9-r137