
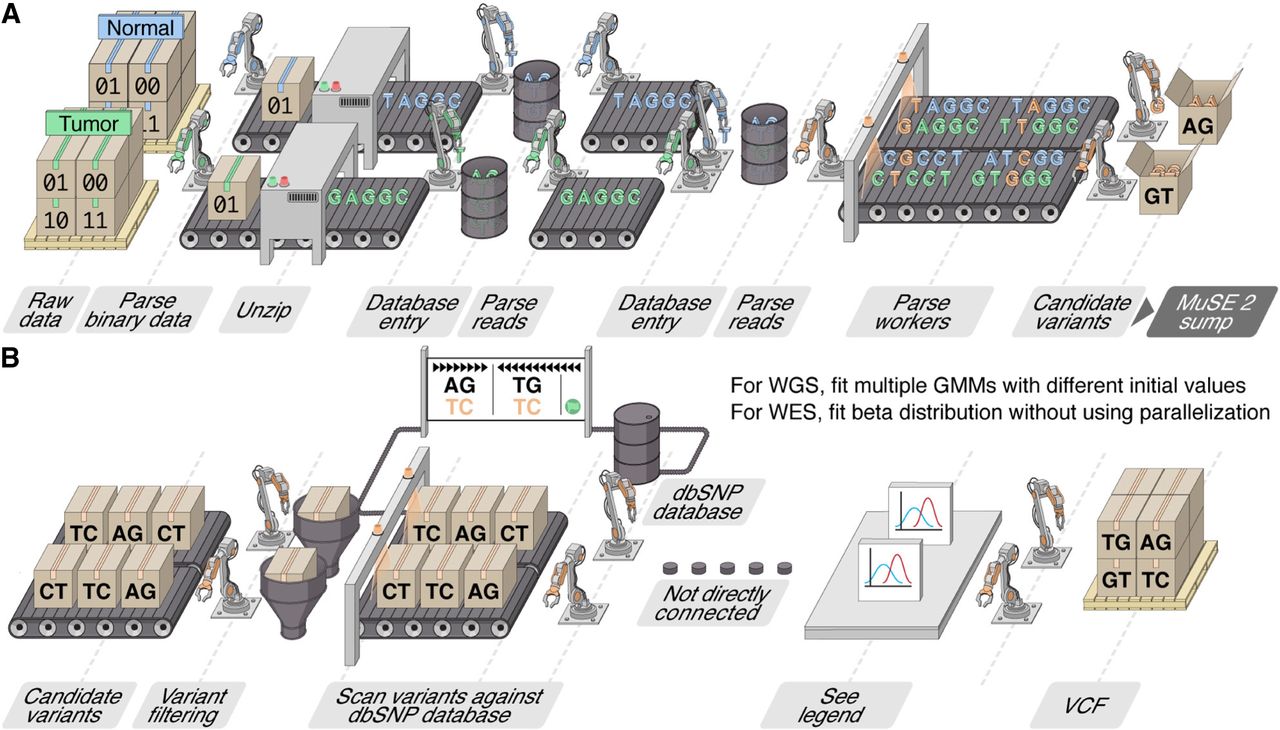
Assembly line illustration of the multistep parallelization implemented in MuSE 2. (A) “MuSE call”: Workers (threads) keep fetching chunks from the input BAM files from the tumor and normal samples and unzipping them to the text format of reads. Downstream workers combine the reads from the tumor and normal samples and send to a queue; from there, other workers detect candidate variants. (B) “MuSE sump”: Multiple workers are used to take the candidate variants and their corresponding estimated summary statistic π’s and scan them against the dbSNP database, labeling those appearing in the database. For candidate variants from the WGS data, we fit two-component Gaussian mixture models (GMMs) with multiple initializations, distributed to multiple workers, in order to separate true variants from background noise; for candidate variants from the WES data, no parallelization is implemented owing to computational simplicity as we simply fit a Beta distribution to π’s.








