
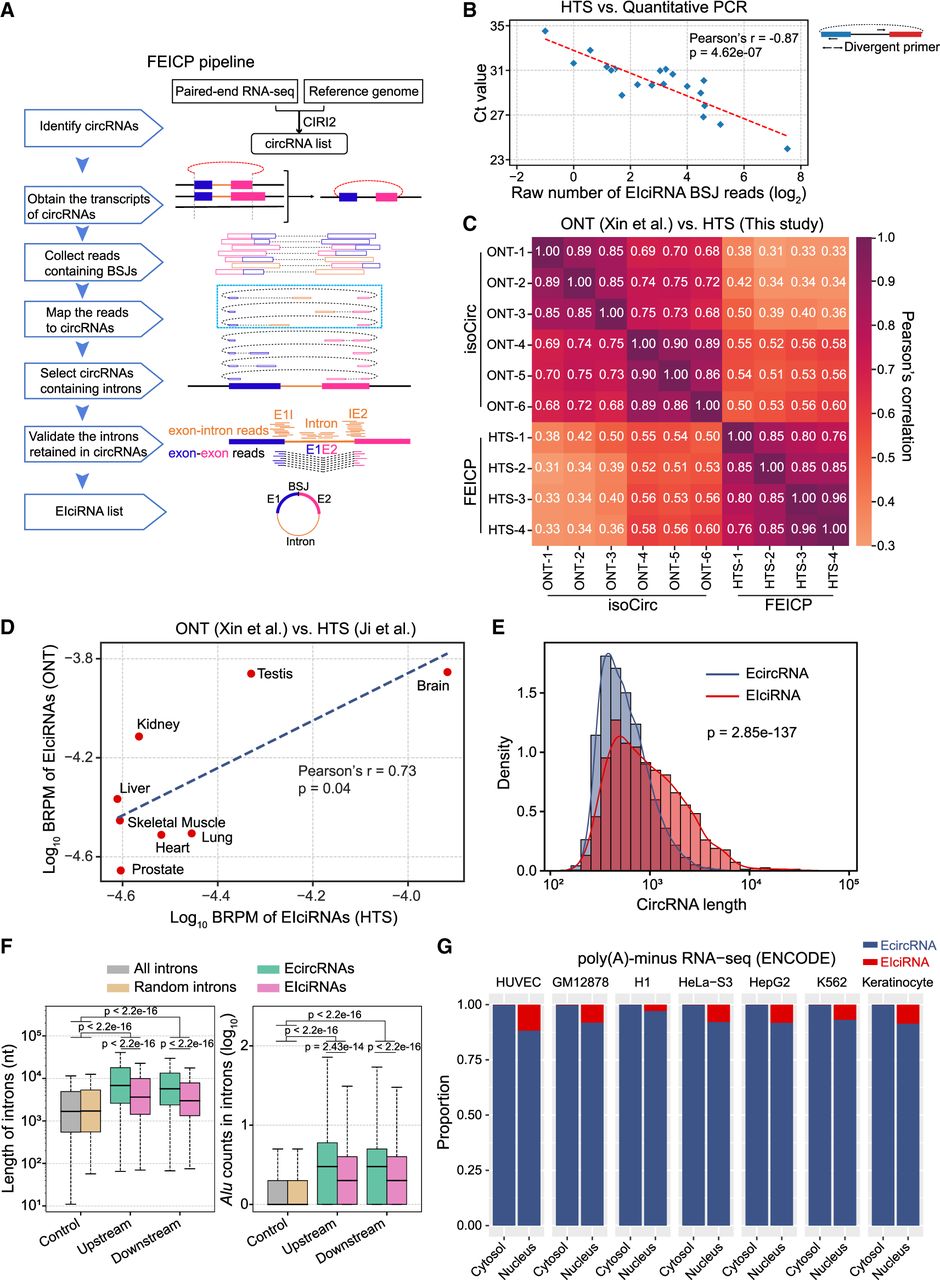
FEICP pipeline and genome-wide analyses of EIciRNAs. (A) Workflow of the pipeline finding EIciRNAs from paired-end RNA sequencing (FEICP). Briefly, circRNAs were identified from paired-end high-throughput sequencing (HTS) data, followed by detecting exon–intron junctions or intron sequences from paired reads of backsplicing junctions (BSJs), and then retained introns and EIciRNAs were validated. (B) Correlation of Ct value examined by RT-qPCR and BSJ reads predicted by FEICP for 20 randomly selected EIciRNAs in HEK293 cells (left). Divergent primers to amplify EIciRNAs were indicated (right). (C) Pearson’s correlation of the BSJ counts of EIciRNAs commonly detected by HTS and Oxford Nanopore Technology (ONT) sequencing. (D) Correlation of EIciRNA BSJ reads (backspliced reads per million [BRPM]) from published HTS or ONT data sets of eight human tissues. (E) Distribution of full-length of EcircRNAs and EIciRNAs in HEK293 cells. The full-length of EcircRNAs and EIciRNAs was calculated by psirc and FEICP, respectively. (F) Boxplots showing the length of flanking introns of EcircRNAs and EIciRNAs (left) and Alu counts in their flanking introns (right). All introns annotated in the human genome and 2000 randomly selected introns were used as controls. (G) Proportion of EcircRNAs and EIciRNAs in cytosol and nucleus across seven human cell lines. For E and F, P-values were calculated using the Wilcoxon rank-sum test.








