
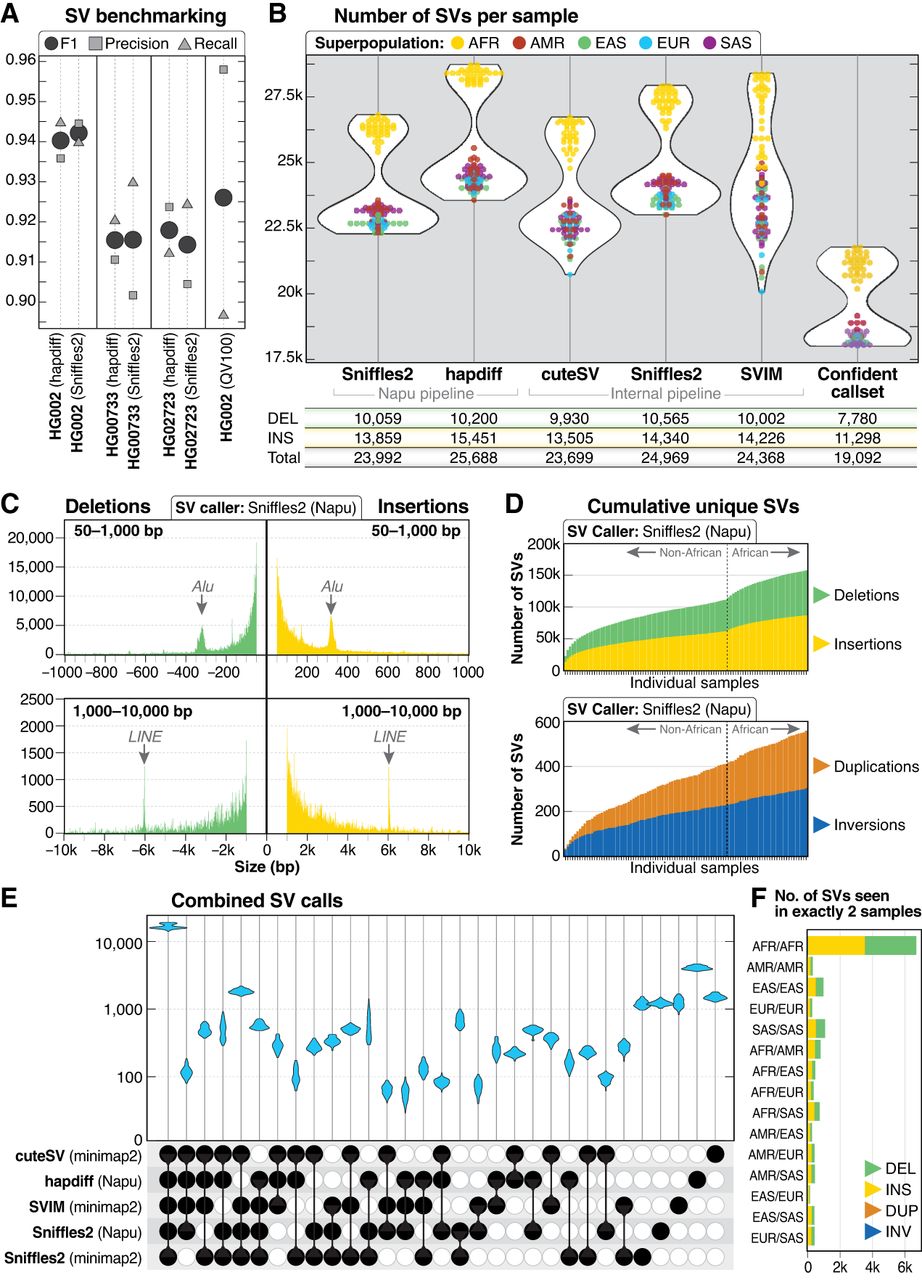
SV call set. (A) SV calls were benchmarked against HPRC Sniffles2 SV calls within the GIAB HG002 SV Tier1 benchmarking regions. (B) A similar number of genome-wide SVs were identified by all five callers used in this study. The confident call set is defined as variants called by hapdiff and at least two unique alignment-based callers. For each call set, the average number of deletions (DEL), insertions (INS), and total SVs (including INV, DUP, and BND events) per sample is shown. (C) Histogram of insertion and deletion counts stratified by size. The peak ∼300 bp represents Alu insertions or deletions, and the peak ∼6 kbp represents LINE insertions or deletions. (D) Cumulative novel SVs per sample. The frequency of new SVs observed increases when samples from individuals of African ancestry are included. (E) Upset plot of overlap among SV callers after merging with Jasmine. For each sample, five VCF files were merged, demonstrating that the majority of calls in each sample were called by all five callers. (F) Among 113,696 SVs from the Jasmine-merged confident call set, 12,432 were found in exactly two samples, with 6181 (50%) of those calls in pairs in which both samples are from the African superpopulation.








