
Long-read subcellular fractionation and sequencing reveals the translational fate of full-length mRNA isoforms during neuronal differentiation
- 1Department of Biomolecular Engineering, University of California Santa Cruz, Santa Cruz, California 95064, USA;
- 2Department of Molecular, Cell, and Developmental Biology and Center for Molecular Biology of RNA, University of California Santa Cruz, Santa Cruz, California 95064, USA
-
↵3 These authors contributed equally to this work.
Abstract
Alternative splicing (AS) alters the cis-regulatory landscape of mRNA isoforms, leading to transcripts with distinct localization, stability, and translational efficiency. To rigorously investigate mRNA isoform-specific ribosome association, we generated subcellular fractionation and sequencing (Frac-seq) libraries using both conventional short reads and long reads from human embryonic stem cells (ESCs) and neural progenitor cells (NPCs) derived from the same ESCs. We performed de novo transcriptome assembly from high-confidence long reads from cytosolic, monosomal, light, and heavy polyribosomal fractions and quantified their abundance using short reads from their respective subcellular fractions. Thousands of transcripts in each cell type exhibited association with particular subcellular fractions relative to the cytosol. Of the multi-isoform genes, 27% and 19% exhibited significant differential isoform sedimentation in ESCs and NPCs, respectively. Alternative promoter usage and internal exon skipping accounted for the majority of differences between isoforms from the same gene. Random forest classifiers implicated coding sequence (CDS) and untranslated region (UTR) lengths as important determinants of isoform-specific sedimentation profiles, and motif analyses reveal potential cell type–specific and subcellular fraction–associated RNA-binding protein signatures. Taken together, our data demonstrate that alternative mRNA processing within the CDS and UTRs impacts the translational control of mRNA isoforms during stem cell differentiation, and highlight the utility of using a novel long-read sequencing–based method to study translational control.
Accurate eukaryotic gene expression requires messenger RNA (mRNA) assembly from precursor transcripts. Protein coding and regulatory sequences (exons) are distributed across expansive precursor mRNA transcripts. The spliceosome excises intervening noncoding sequences (introns) from pre-mRNA and ligates the exon sequences together to generate translation-competent mRNA (Wang et al. 2015). Conserved sequences at exon–intron boundaries (splice sites) direct spliceosome assembly on each newly synthesized intron. The spliceosome can assemble different combinations of exon sequences to generate mRNA isoforms from a common pre-mRNA transcript (Konarska 1998; Wu et al. 1999). Alternative splicing (AS) generates isoforms not only with distinct protein coding potential but also with different post-transcriptional regulatory capacity. For example, AS decisions that introduce premature termination codons (PTCs) induce nonsense-mediated decay (NMD), whereas other splicing events generate transcripts with distinct subcellular localization or translational control. In addition to generating alternative isoforms with unique coding sequences (CDSs), AS can produce isoforms that differ only in their untranslated regions (UTRs). Elements in the UTRs of mature mRNA play pivotal roles in post-transcriptional regulation. In the 5′ UTR, regulatory sequences like upstream open reading frames (uORFs) and internal ribosome entry sites (IRESs) influence translation initiation efficiency (Morris and Geballe 2000; Hellen and Sarnow 2001; Weber et al. 2023). The 3′ UTR contains various elements such as microRNA binding sites and RNA-binding protein (RBP) recognition sites that modulate mRNA stability, localization, and translation (Ciolli Mattioli et al. 2019; Mayya and Duchaine 2019). Regulatory elements in the CDS can also influence the fate of mRNAs. For example, the RBP ELAVL1 stabilizes target mRNAs by binding to AU-rich elements (AREs) within the CDS, preventing their degradation. Conversely, RBPs like ZFP36 can promote mRNA degradation by binding to AREs in coding regions, leading to mRNA decay (Otsuka et al. 2019). Proteins like IGF2BP1 can bind to coding region instability determinants in the CDS of target mRNAs to enhance their stability (Weidensdorfer et al. 2009). By and large, AS confers complex and multidimensional consequences to the fate of mRNAs through shaping the cis-regulatory landscape of alternative isoforms (Castle et al. 2008; Pan et al. 2008; Wang et al. 2008).
Isoform-specific cytosolic fates are evidenced by poor correlation between steady-state mRNA and protein levels in eukaryotic systems (Lian et al. 2001; Griffin et al. 2002; Cox et al. 2005; Schmidt et al. 2007; Fu et al. 2009). And although factors like mRNA stability and translation initiation efficiency play a role in this disparity, the influence of AS on translational control is often overlooked. A number of methods exist to study translational control, which is the regulatory mechanism in eukaryotic cells that governs the efficiency and timing of protein synthesis from mRNA. A well-established method called Ribo-seq offers genome-wide insights into ribosome occupancy and translation dynamics by capturing single-nucleotide-resolution ribosome footprints, but it can be vulnerable to artifacts and signal biases (Ingolia et al. 2009). RNC-seq captures ribosome nascent-chain complex-bound mRNAs to characterize the translatome, but it does not provide ribosome footprints or ribosome density information (Wang et al. 2013). TRAP-seq utilizes epitope-tagged ribosomes to enable cell type–specific translation profiling, which generates data similar to RNC-seq, which can be modified to produce ribosome footprints, but it relies on transgenic models and may not fully replicate endogenous ribosome behavior (Heiman et al. 2014; Reynoso et al. 2015). Frac-seq, which our proposed method builds on, isolates actively translating ribosomes and assesses translation efficiency by stratifying transcripts by the number of ribosomes with which they are associated (Sterne-Weiler et al. 2013). However, it has the potential for selective bias toward highly abundant transcripts, and it lacks single-nucleotide resolution of ribosome positions on mRNA. Although each method has its strengths and weaknesses, one shared disadvantage is that they all involve the sequencing of short mRNA fragments from ribosome-protected or ribosome-associated mRNAs.
Short-read RNA sequencing methods struggle to accurately capture the complete structures of complex RNA isoforms (Steijger et al. 2013). In contrast, long-read RNA sequencing provides full-length reads that span entire transcripts, enabling precise characterization of intricate isoforms and annotation-agnostic detection of novel structures. The primary shortcoming of long-read sequencing is its relatively lower throughput compared with short-read sequencing platforms, limiting the depth of coverage for a given budget. To address this limitation and to maximize the benefits of both long-read and short-read methods, we employed a complementary approach. Here we introduce the development of long-read Frac-seq to obtain full-length transcript isoforms with intact records of ribosome association, structural variation, and long-range interactions. We complement these data with short-read Frac-seq to compensate for the loss of throughput and to provide a more complete and accurate representation of the translated transcriptome.
Results
Characterization of a transcriptome supplemented with long-read-derived novel transcripts
To investigate the relationship between alternative pre-mRNA splicing and isoform-specific mRNA translation, we capitalized on the capability of long-read sequencing to capture complete transcript structures of polyribosome-associated mRNA, without sacrificing throughput by generating both long-read and short-read Frac-seq libraries (Sterne-Weiler et al. 2013). We used human embryonic stem cells (ESCs) and neural progenitor cells (NPCs) as a model system to characterize the translated transcriptome during early neuronal differentiation. The resulting samples were the cytosol, monosome (Mono), light polyribosome (LPR; two to four ribosomes), and heavy polyribosome (HPR; five or more ribosomes) fractions (Fig. 1A). By utilizing the R2C2 method (Volden et al. 2018), our long-read libraries, with a mean read length of 2 kb and a mean library size of 500,000, were reinforced with improved base-calling accuracy (93%) and with high-confidence transcript starts and ends. Fractionation of the long reads was employed to enhance the likelihood of detecting transcripts that may be preferentially associated with distinct subcellular fractions. All long-read libraries were pooled for de novo transcriptome assembly using Mandalorion (Volden et al. 2023). Mandalorion assembled a transcriptome consisting of transcripts with long-read support of five or more. We then used SQANTI3 to quality control and filter the initial transcriptome, resulting in a long-read-derived transcriptome containing only those transcripts with 20 or more total mean normalized short-read counts across all fractions for each cell type. To this, we applied functional annotation using all three modules of the Functional IsoTranscriptomics analysis suite (de la Fuente et al. 2020). The resulting long-read-derived transcriptome was then merged with GENCODE's GRCh38.p13 release 41 primary assembly annotation (Harrow et al. 2012) to account for transcripts that were not captured by long-read sequencing. The following analyses were done in the context of this “comprehensive transcriptome” containing both annotated and long-read-derived novel transcripts.
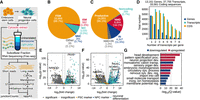
Experimental overview and characterization of the comprehensive transcriptome and the cytosol. (A) Schematic of the experiment and subsequent bioinformatic analysis workflow of the resulting cytosolic extract and fractionated, ribosome-associated short and long reads from ESCs and NPCs. (B) The transcriptome classified by SQANTI3-defined structural categories of spliced transcripts, including: full splice match (FSM), incomplete splice match (ISM), novel in catalog (NIC), novel not in catalog (NNC), and intergenic or fusion transcripts (other). FSM and ISM transcripts match annotated splice sites and junctions (in GRCh38.p13 release 41), whereas NIC transcripts comprise novel combinations of annotated splice sites and junctions, and NNC transcripts contain at least one unannotated splice site. (C) The transcriptome classified by productivity based on the detection of complete or incomplete open reading frames (productive or noncoding respectively), premature stop codons (NMD), and retained introns (RIs). (D) Stratification of the transcriptome by the number of isoforms and unique coding sequences per gene. (E,F) Gene-level (E) and transcript-level (F) differential expression between NPC and ESC cytosolic fractions. (G) Top 12 enriched Metascape pathways in differentially expressed genes between NPC and ESC cytosolic fractions.
The comprehensive transcriptome had short-read coverage meeting a one count per million reads (CPM) cutoff for 37,755 transcripts with 33,561 unique CDSs, arising from 13,161 genes (Fig. 1D). Of these, 5875 and 4590 transcripts from 4095 and 3176 genes were uniquely expressed in ESCs or NPCs, respectively. Transcripts were organized into SQANTI3-defined structural categories based on their fidelity to transcript structures in the GRCh38.p13 release 41 primary assembly annotation (Harrow et al. 2012): 91.2% of transcripts matched the annotation; 8.7% were considered novel (containing either novel combinations of known splice sites and junctions or at least one novel splice site); and 0.1% were categorized as either genic or fusions (Fig. 1B). Additionally, transcripts were categorized based on their productivity. We define productive transcripts as those encoding a full-length, canonical protein. Unproductive classes include noncoding (lacking a complete open reading frame), NMD, and retained intron (RI): 66.1% of transcripts were considered productive; 0.7% were predicted to be noncoding; 18.1% were classed as NMD based on the presence of a PTC; 11.5% had a RI; and the remaining 3.6% met both NMD and RI conditions (Fig. 1C).
We used Salmon (Patro et al. 2017) to pseudoalign the fractionated short reads, with an average library size of 71.5 million reads, to the comprehensive transcriptome, producing transcript-level quantification across the gradient. Using the cytosolic fraction, which represents the raw output of the nucleus, we next tested the baseline transcriptomic differences in NPCs relative to ESCs at the gene level (Fig. 1E) and at the transcript level (Fig. 1F) to reveal upregulation of NPCs and neuronal differentiation markers and downregulation of pluripotency markers. Twenty-two and 24 transcripts from genes that are PSC or NPC markers or that are associated with neuronal differentiation had distinct ribosome association profiles in ESCs and NPCs, respectively. Only three of those transcripts had distinct ribosome association profiles in both cell types, and they all associate with the Mono regardless of expression differences. In these few examples, we did not observe a compensatory relationship between ribosome association and expression levels. Metascape (Zhou et al. 2019) pathways further encapsulated these observations (Fig. 1G). Taken together, these results present the framework for an approach to integrate fractionated long and short reads to study translational control at isoform-level resolution.
Thousands of transcripts exhibit distinct association with particular subcellular fractions
To discover if mRNA transcripts have distinct ribosome association profiles, we clustered transcript-level expression trajectories across the gradient using tappAS (de la Fuente et al. 2020), revealing subpopulations of transcripts with clear enrichment in one subcellular fraction over the others (Fig. 2A; Supplemental Tables 4, 5). A subpopulation of transcripts enriched in both the Mono and LPR fractions stood out as one of the most populous subsets, making up ∼30% of transcripts with enrichment in subcellular fractions in both cell types. Thus, subsequent analyses class Mono-associated transcripts and LPR-associated transcripts as those that are exclusively enriched in those fractions, leaving the set of Mono and LPR-associated (M + L) transcripts as a standalone subpopulation. Thousands of transcripts were considered significantly enriched (log2FC ≥ 1.0, P-value ≤ 0.05) in subcellular fractions relative to the cytosol (Fig. 2B). Overall, 7.5% and 6.8% of transcripts were significantly associated with a subcellular fraction in ESCs and NPCs, respectively. In the context of nonmutually exclusive enrichment in subcellular fractions, subpopulations of transcripts were generally dissimilar across fractions within cell type, with the exception of the Mono and LPR fractions with Jaccard similarity of 0.41 and 0.33 in ESCs and NPCs, respectively (Supplemental Fig. 1), owing to the substantial M + L transcript subpopulations. When stratified by productivity, Mono- and LPR-associated transcript subpopulations exhibited pronounced incorporation of unproductive classes relative to the cytosol. HPR-associated transcripts displayed a relative reduction of unproductive classes relative to the cytosol in ESCs, whereas an increase is observed in NPCs (Fig. 2C). These findings support the hypothesis that levels of ribosome association may correlate with levels of translatability.
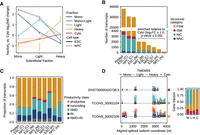
Establishing transcript ribosome association profiles. (A) Clustering of transcripts by their expression trajectories across the gradient relative to the cytosol. (B) Extraction of fraction-associated transcripts based on significant enrichment (log2FC ≥ 1.0, P-value ≤ 0.05) in the Mono, LPR, or HPR fractions relative to their abundance in the cytosol. Fractions: (C) cytosol, (M) Mono, (L) LPR, and (H) HPR. (C) Categorization of fraction-associated transcripts by productivity. (D) Differential sedimentation of three isoforms in TMEM59. Above spliced isoform models, histograms of short-read support at exons are colored by fraction. The stacked barplot summarizes the proportion of total gene expression each isoform contributes in each subcellular fraction.
AS confers functional consequences to the stability and translation of mRNAs
Because we observed transcript subpopulations with distinct ribosome association profiles, we postulated that alternative mRNA isoforms may likewise sediment discretely. To test this hypothesis, we calculated the expression of individual isoforms relative to all isoforms from the same gene and measured the difference between their gene fractions in subcellular fractions relative to those in the cytosol (Supplemental Tables 6, 7). TMEM59 is an example of a gene with three isoforms, two of which exhibit differential sedimentation in ESCs (Fig. 2D). TMEM59 expression in ESCs is composed of a M + L-associated isoform, an HPR-associated isoform, and a cytosol-associated (not differentially sedimenting) isoform. Endogenous post-transcriptional silencing of TMEM59 by miR-351 in murine neural stem cells has been implicated to promote neuronal differentiation (Li et al. 2012), although the two differentially sedimenting isoforms share all but the last base of their 3′ UTRs. But along similar lines, it may be the case that cis-regulatory differences in their 5′ UTRs and CDSs influence the isoform-specific nature of their sedimentation.
We found 3321 (26.5%) and 2254 (19.2%) genes in ESCs and NPCs, respectively, exhibiting differential isoform sedimentation (|Δ gene fraction| ≥ 0.1 and Q-value ≤ 0.05) in a subcellular fraction relative to the cytosol (Fig. 3A). Within those genes, 4906 and 3229 transcript isoforms preferentially sedimented (Δ gene fraction ≥ 0.1) with a subcellular fraction in ESCs and NPCs, respectively. These instances substantiate AS as an architect of isoform-specific translational control. We observed decreasing concordance of gene fraction changes across the gradient between cell types, with a Pearson correlation coefficient of 0.59, 0.54, and 0.09 in Mono, LPR, and HPR, respectively. In fact, 3085 transcripts in 1506 genes exhibit divergent patterns of isoform sedimentation (Supplemental Fig. 2). Together, these findings suggest that isoform-specific sedimentation is likely cell type specific, possibly owing to differences in the composition and environment of trans-acting factors.
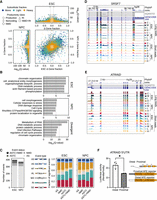
Differential isoform sedimentation across the gradient and functional outcomes of alternative splicing (AS). (A) Volcano plots representing differential isoform sedimentation by changes in isoform gene fraction relative to the cytosol; 3321 and 2254 genes in ESCs and NPCs, respectively, exhibit significant differential isoform sedimentation (|Δ gene fraction| ≥ 0.1, Q-value ≤ 0.05). The central plot shows changes in isoform gene fraction, with Q-value ≤ 0.05, of isoforms expressed in both ESCs and NPCs. (B) The first two bar plots show the top six enriched Metascape pathways in genes containing significant instances of differential isoform sedimentation exclusively in ESCs or NPCs. The third bar plot, labeled “divergent,” depicts enriched Metascape pathways in genes displaying contrasting patterns of isoform sedimentation between ESCs and NPCs. (C) The first stacked bar plot categorizes significant AS events (|ΔΨ| ≥ 0.1, adjusted P-value or Q-value ≤ 0.05) as AS; AS coupled with translational control (ASTC), meaning splicing events that are differentially included across the gradient; NMD; and AS coupled with both translational control and nonsense-mediated decay (ASTC + NMD). The following two bar plots show the breakdown of event types comprising each category in ESCs and NPCs. (D,E) UCSC Genome Browser snapshot of long-read and short-read coverage at SRSF7 (D), exhibiting subcellular fraction–associated inclusion of a conserved retained intron, and at ATRAID (E), exhibiting subcellular fraction–associated alternative first exon usage. Fractions: (C) cytosol, (M) Mono, (L) LPR, and (H) HPR. (F) Luciferase assay measuring the translational impact of using either the distal or the proximal ATRAID 5′ UTR in HEK293 cells.
Additionally, Figure 3A illustrates that isoforms associated with the Mono and LPR fractions have a greater magnitude of gene fraction differences than the HPR fraction relative to the cytosol. This finding suggests that the Mono, LPR, and HPR fractions gradate toward increasing similarity in transcript abundance and isoform ratios with the cytosol, which is consistent with findings from other groups (Floor and Doudna 2016). Considering these results, we posit that the average number of ribosomes per mRNA in the cytosolic fractions of our ESC and NPC samples may be similar to that of the HPR fraction. Among genes displaying differential isoform sedimentation, pathways involved in chromatin organization, organophosphate biosynthesis, and phosphorylation were enriched in ESCs specifically, whereas DNA damage, stress response, and cell cycle pathways were enriched in NPCs (Fig. 3B). Genes demonstrating divergent isoform sedimentation across cell types were enriched in similar pathways, with the addition of RNA metabolism. When comparing the Gene Ontology of cognate subpopulations of subcellular fraction–associated transcripts across cell types, we observed that cell cycle, translation, and mRNA processing–related pathways were consistently represented, whereas cell type–specific pathway enrichment was much more apparent in the cytosolic fraction (Supplemental Fig. 3).
To examine the types of AS that give rise to the diversity of the transcriptome, we categorized AS events as AS (0.1 ≤ Ψ ≤ 0.9, adjusted P-value ≤ 0.05 within condition), ASTC (|ΔΨ| ≥ 0.1, Q-value ≤ 0.05 across subcellular fractions), NMD (events that introduce a PTC), and ASTC + NMD (NMD events that adhere to the mentioned cutoffs for significant ASTC events) (Fig. 3C; Supplemental Tables 8, 9); 11.8% and 7.0% of significant AS events were classified as either ASTC or ASTC + NMD in ESCs and NPCs, respectively. We found that alternative first exon, RI, and skipped exon (SE) events feature most prominently among ASTC events, whereas SEs and RIs comprise the majority of ASTC + NMD events; 2456 and 1257 CDS-altering events (A3, A5, MS, MX, and SE event types) and 526 and 359 terminal events (AF and AL event types) were linked to translational control (either ASTC or ASTC + NMD) in ESCs and NPCs, respectively. Because of the mentioned similarity between the HPR fraction and the cytosol, the majority of ASTC/ASTC + NMD events were Mono (79.1% in ESCs, 62.1% in NPCs) and LPR associated (42.5% in ESCs, 54.8% in NPCs).
In our data set, SRSF7 contained one complete and one partial RI event associated with NMD via induction of a PTC in the highly conserved SRSF7 intron 3 locus, which has been previously described to contain a conserved poison exon (Fig. 3D; Lareau et al. 2007; Königs et al. 2020). Preferential association of PTC-containing isoforms with the Mono, as well as modestly with the LPR fraction, is consistent with our understanding of NMD's effect on translation (Maquat et al. 1981; Celik et al. 2017; Nickless et al. 2017). ATRAID, a relatively poorly understood gene implicated to play roles in all-trans retinoic acid–induced apoptosis, osteoblast differentiation, and some cancer types (Ding et al. 2015; Zhang et al. 2023), demonstrates marked patterns of alternative first exon usage across the gradient (Fig. 3E). The proximal first exon of ATRAID was preferentially spliced into the Mono-associated isoform, which may indicate its reduced translation. Indeed, a Renilla-firefly luciferase assay comparing Renilla incorporating either the proximal or the distal 5′ UTR of ATRAID in HEK293 cells exhibited significant differences in fluorescence (Fig. 3F). The two isoforms of ATRAID may potentially be functionally different, as the distal first exon contains an uORF, which may encode a signal peptide with potential importance to its localization with lysosomes (Ding et al. 2015). Collectively, these results demonstrate the widespread functional impacts of AS to the cytosolic fate of mRNAs.
Intrinsic features and cis-elements correlate with transcript polyribosome profiles
Given that AS defines the cis-regulatory landscape of mature mRNAs, we sought to identify intrinsic transcript features that may encode the underlying regulatory grammar of ASTC. We define intrinsic transcript features as measurements and functional elements that are native to the sequence of a spliced transcript. To extract the relative predictive weight of features on ASTC, we employed random forest classifiers (RFCs) to perform feature selection. Across the transcriptome, we measured the length and GC-content of the transcript, CDS, and UTRs. Additionally, our feature set included 5′ and 3′ UTR motifs, uORFs, repeat sequences, coding capacity, codon frequency, the presence of PTCs, and RIs (Supplemental Table 3). Given these features, RFCs were assigned binary classification tasks to predict the correct subcellular fraction for transcripts between every combination of subcellular fraction–associated transcript subpopulations at an 80:20 train:test split using 300 estimators. From the results, we extracted permutation feature importance, with 50 repeats, and found that the number of exons, length of the CDS and UTRs, and the GC-content of the UTRs were important features for our models to correctly classify transcript polyribosome profiles. We note, however, that although our RFC models outperformed unskilled models/chance levels, the combination of generally adequate receiver operating characteristic curves with suboptimal Precision-Recall performance highlights class imbalances and the need for more data points in each fraction and further indicates the requirement of additional features beyond those included in this study (Supplemental Fig. 4).
Our findings that CDS and 3′ UTR length positively correlate with association with heavier polyribosome fractions is consistent with previous reports (Fig. 4A; Floor and Doudna 2016). This is not to be confused with ribosome density, which other groups have shown to be inversely correlated with CDS length (Arava et al. 2003; Zhao et al. 2017). Although a longer CDS can theoretically accommodate a greater number of ribosomes, the increased potential for incorporation of nonoptimal or rare codons may trigger codon usage-dependent negative impacts to translation initiation and elongation (Lyu et al. 2021). Additionally, longer CDS and transcript lengths have been observed to be negatively correlated with translation initiation rates in the context of intra-polysomal ribosome reinitiation (Rogers et al. 2017). Also, although CDS length and 3′ UTR length, individually, positively correlate with ribosome association, we did not observe clear instances of linkage between SE and AL event inclusion. En masse, inference of ribosome association based on the CDS alone is likely too simplistic to make accurate predictions.
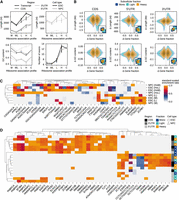
Analysis of features correlated with ribosome association profiles. (A) The number of exons and summaries of length and GC-content, with 90% confidence interval, of the CDS, 5′ UTR and 3′ UTR of transcripts associated with subcellular fractions. Fractions: (C) cytosol, (M) Mono, (ML) Mono + Light, (L) LPR, and (H) HPR. (B) Measurement of the change in isoform gene fraction relative to the cytosol and differences in CDS, 5′ UTR and 3′ UTR length, and GC-content of differentially sedimenting isoforms relative to the dominant isoform in the cytosol. Kernel densities for all coding isoforms are drawn with a 0.2 threshold. Subplots in the bottom left of each plot summarize the relative abundance of observations in each quadrant of their respective main plot, colored by fraction. (C) HOMER-derived de novo sequence motif and known RBP motif enrichment ratios in skipped exons enriched in subcellular fractions versus skipped exons not enriched in each given fraction. “M” and “L” refer to the Mono and LPR-associated FASE sets, whereas “HvM” and “HvL” refer to the HPR-associated FASE sets relative to the Mono and LPR fractions, respectively. (D) Enrichment of motifs using the same approach as in C, but in 30 nt windows of the CDS, 5′ UTR, and 3′ UTR of isoforms exhibiting divergent sedimentation profiles across cell types. The target sets were made from isoforms that preferentially sediment with each given subcellular fraction in one cell type, and the background sets were made from isoforms exhibiting the same in the other cell type. The standard-scaled enrichment ratio colorbar is shared by C and D.
To more clearly understand changes in feature length that may impact ribosome association, we also measured the change in CDS, 5′ UTR, and 3′ UTR length relative to the dominant cytosolic isoform among isoforms belonging to genes with differentially sedimenting isoforms (termed, gene-linked isoforms). Mono- and LPR-associated isoforms displayed a clear signal of relatively shorter CDS and longer 3′ UTR, whereas HPR-associated isoforms remained largely similar or equivalent to the dominant cytosolic isoform (Fig. 4B). Relatively longer 3′ UTRs in gene-linked isoforms are connected to strong effects on ribosome association, and isoforms with 5′ UTRs ≥ 1000 nt in length have been observed to be relatively poorly ribosome associated relative to their shorter 5′ UTR-containing counterparts within the same gene (Floor and Doudna 2016). These phenomena could be owing, in part, to the potential increased inclusion of cis-regulatory elements in UTRs including miRNA target sites, uORFs, and iron-responsive elements, which can negatively impact mRNA stability and translation. Our summary analyses of GC-content measurements yielded less clear patterns in relation to ribosome association (Fig. 4B). Nonetheless, ∼30% of isoforms preferentially sedimenting in lowly ribosome-associated fractions (Mono and LPR) exhibited decreasing 3′ UTR GC-content relative to the dominant cytosolic isoform, which may support findings that relate lower 3′ UTR GC-content to increased association with P-bodies and enhanced susceptibility to miRNA targeting (Courel et al. 2019). The other 70% of lowly ribosome-associated isoforms showed the opposite characteristic regarding 3′ UTR GC-content, which is concordant with reports that suggest an inverse relationship between 3′ UTR GC-content and mRNA stability (Litterman et al. 2019). Overall, broad measurements like length and GC-content were predictive of ribosome association to some degree but appear to lack the granularity required to definitively elucidate the mechanisms underlying instances of ASTC. Comparisons between cell types regarding feature measurements also reveals cell type–specific differences in trends that indicate further layers of complexity (Supplemental Figs. 5, 6).
To look beyond length and GC-content measurements, we identified sequence motifs that are associated with subcellular fractions. To do this, we took fraction-associated skipped exons (FASEs), exons that were determined to be significantly enriched in a subcellular fraction (ΔΨ ≥ 0.1, Q-value ≤ 0.05 across subcellular fractions) relative to the cytosol, and sliced them into 30 nt windows. Each set of windows was complemented with a background set consisting of windows made from SEs that were not significantly enriched in their given subcellular fraction. The HPR fraction was tested for enrichment against the Mono and LPR fraction (HvM and HvL, respectively) to produce HPR FASE sets owing to a dearth of HPR-enriched exons relative to the cytosol. The resulting sets of FASEs included about 415 and 240 exons on average for each subcellular fraction in ESCs and NPCs, respectively.
Using HOMER (Heinz et al. 2010) on each set of windows, we discovered 111 de novo motifs, in total, that were significantly enriched (P-value ≤ 0.05, FDR ≤ 0.2) in the target sequences over their respective background sets. We used Tomtom (Gupta et al. 2007) to identify the best matches (P-value ≤ 0.05) between the de novo motifs and known RBP motifs in the Homo sapiens data set (Ray et al. 2013). We next combined the set of motifs with the CISBP-RNA H. sapiens RBP motif set (Ray et al. 2013) and used SEA (Bailey and Grant 2021) to measure their enrichment (P-value ≤ 0.05, enrichment ratio ≥ 1.1) in each set of FASEs (Fig. 4C; Supplemental Table 10). Several motifs exhibited enrichment in at least one fraction, with motif enrichment bisecting into clusters of Mono and LPR FASE sets and of HPR-associated FASE sets regardless of cell type.
We applied the same approach to the CDS, 3′ UTR, and 5′ UTR of divergently sedimenting isoforms between ESCs and NPCs to identify cell type–specific motifs that may underlie the differences in their sedimentation (Supplemental Table 11). Target sequences were generated from isoforms preferentially sedimenting with each given fraction in one cell type versus those preferentially sedimenting with the same fraction in the other cell type. Motif enrichment in these sets of sequences clustered more distinctly by cell type than by fraction, and most motifs were exclusively enriched in one cell type and not the other. We acknowledge, however, that motif analyses are limited by the fact that RBP binding specificities are often multivalent and difficult to predict. Nonetheless, we report the presence of statistically significant sequence motifs enriched in FASEs, as well as those that are differentially enriched and utilized between divergently sedimenting isoforms. Altogether, 19 of the 43 known RBPs identified as fraction or cell type specific have been previously implicated in translational control or observed to associate with polyribosomes (Supplemental Tables 10, 11; Paronetto et al. 2006; Smart et al. 2007; Markus and Morris 2009; Jin et al. 2011; Svitkin et al. 2013; Aviner et al. 2017; Ueno et al. 2019; Rizzotto et al. 2020; Sévigny et al. 2020; Zhang et al. 2021; Anisimova et al. 2023). Additional studies are necessary to test the role of these potential factors in ASTC. Because we were specifically interested in motifs related to ribosome association, we did not perform motif analysis on introns. As a whole, these results suggest that ribosome association is impacted by the composition of intrinsic transcript features, likely with combinatorial effects.
Discussion
Here, we report the first integration of long-read RNA sequencing with a translatomic method, which we call LR Frac-seq, and we describe an approach to integrate long-read and short-read Frac-seq to characterize the translated transcriptome in human ESCs and NPCs. We took a complementary approach to capitalize on the major strength of long-read sequencing in capturing complete transcript structures, while leveraging short-read sequencing's significantly higher throughput for accurate quantification. Many examples of hybrid sequencing approaches have previously been applied to complex biological problems by other groups (Puglia et al. 2020; Leshkowitz et al. 2022; Reese et al. 2023). For the long reads, we employed the R2C2 method to generate high-confidence consensus sequences with high base-calling accuracy and well-defined transcript start and end sites. With these, we performed de novo transcriptome assembly to generate the set of full-length transcripts detected in the system, deemed the long-read-derived transcriptome. Indeed, the long-read-derived transcriptome does not comprehensively capture the entirety of the expressed transcriptome in ESCs and NPCs, as indicated by short-read transcript-level mapping rates: On average, 87% of short reads mapped to the genome, whereas 42% mapped to the long-read-derived transcriptome (Supplemental Table 1). The high quality of the short reads and the high mapping rate of the long reads to the genome (Supplemental Table 2) suggest that the lower short-read transcriptomic mapping rate is owing to the incomprehensive nature of the long-read-derived transcriptome, which can likely be improved by deeper sequencing; ideally at 1 million or greater reads per long-read library. To account for transcripts potentially missed by long-read sequencing, we merged the long-read-derived transcriptome annotation with GENCODE's GRCh38.p13 release 41 primary assembly annotation (Harrow et al. 2012) to produce a nonredundant, “comprehensive” transcriptome annotation for downstream analyses. The much deeper fractionated short-read libraries were utilized to quantify the comprehensive transcriptome across the gradient, consisting of the cytosol, Mono, LPR (two to four ribosomes), and HPR (five or more ribosomes) fractions. Highlighting one of the major benefits of long-read sequencing, we found 3281 transcripts with either novel combinations of known splice sites or one or more novel splice sites, accounting for 8.7% of the expressed (≥1 CPM) comprehensive transcriptome.
We compared transcript abundances in subcellular fractions to their cognate cytosolic fractions to identify transcripts with enrichment in particular fractions relative to the cytosol, which represent the raw output of the nucleus. We found that 7.5% and 6.8% of transcripts, in ESCs and NPCs, respectively, preferentially associate with subcellular fractions (Fig. 5A,C) and that the proportion of productive transcripts associated with a given fraction directly correlates with ribosome association (Fig. 5B,D). Isoforms observed to preferentially sediment in subcellular fractions accounted for 13% and 9.8% of transcripts in multi-isoform genes (Fig. 5A,C). We trained RFCs to select features at the transcript level, and we found that the number of exons and the CDS, 5′ UTR, and 3′ UTR length along with 5′ UTR and 3′ UTR GC-content were the most important features in our feature set for the accurate prediction of transcript polyribosome profiles in our data set.
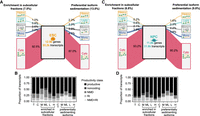
Summary of transcriptomic ribosome association profiles. (A,C) We identified thousands of transcripts in ESC and NPC whose expression was significantly higher (log2FC ≥ 1.0, P-value ≤ 0.05) in a subcellular fraction relative to the cytosol. We also identified thousands of isoforms in multi-isoform genes whose gene fraction was significantly higher (Δ gene fraction ≥ 0.1, Q-value ≤ 0.05) in a subcellular fraction relative to its gene fraction in the cytosol. (B,D) The proportion of productive and unproductive transcript classes in each subpopulation of transcripts enriched and/or preferentially sedimenting in subcellular fractions. (T) Whole expressed transcriptome in the given cell type. Fractions: (C) cytosol, (M) Mono, (ML) Mono + Light, (L) LPR, and (H) HPR. (C*) The subset of transcripts comprising multi-isoform genes specifically, in the cytosol.
Among multi-isoform genes expressed in both ESCs and NPCs, gene-linked differences in isoform sedimentation relative to the cytosol were largely cell type specific, although patterns of intrinsic transcript feature differences between fractions were similar between cell types (Supplemental Figs. 2, 5, 6). We found that isoforms with a shorter CDS and longer 3′ UTR relative to the dominant isoform in the cytosol corresponded most clearly to the Mono and LPR sedimentation profiles. Additionally, motif analyses revealed potential RBP motifs in FASEs and in divergently sedimenting isoforms. These motifs cluster by fraction and by cell type, respectively (Fig. 4C,D). In total, binding sites for 43 unique RBPs exhibited fraction-specific enrichment, and nearly half (44%) have previously established roles in translational control or demonstrated association with polyribosomes (Supplemental Tables 10, 11). For example, proteomic analysis of polyribosomes revealed numerous splicing factors, including HNRNPC, SRSF10, and SRSF7 as polyribosome associated (Aviner et al. 2017). Many of these factors have distinct sedimentation profiles across sucrose gradients, an observation that is consistent with the fraction-specific enrichment of RBP binding sites observed here. As a whole, our results present intrinsic feature measurements and potential RBP motifs that likely enact combinatorial effects on translation, providing both previously reported and novel insights into the underlying mechanisms of ASTC. Because the most predictive intrinsic features were rather broad, we hypothesize that inter-isoform differences in length and GC-content more likely vaguely encapsulate changes to the isoform-specific cis-regulatory landscape. Several factors may affect an mRNA's translational output, including intrinsic and trans-acting influences to mRNA stability, post-transcriptional modifications, and combinatorial interactions with multiple RBPs. Therefore, it may be difficult to distill trends in transcript-level features across polyribosome fractions without also measuring transcriptome-wide mRNA half-life and capturing RBP–mRNA interactions, for example.
Because the LPR and HPR fractions were pooled sets of individual polyribosome fractions, we could not assess features in the context of ribosome density. To be clear, LR Frac-seq can be performed without pooling individual polyribosome fractions, which would enable analyses at the level of ribosome density. We note that the HPR fraction is likely composed of both efficiently and inefficiently translated transcripts depending on their ribosome density and that trends of feature length and GC-content are subject to exceptions in each subcellular fraction. Additionally, Frac-seq differs from ribosome profiling methods in that it does not capture single-nucleotide resolution ribosome footprints. Rather, it stratifies the translated transcriptome in terms of the number of ribosomes associated with full-length mRNAs. Therefore, it is not intended to replace ribosome profiling methods and is instead an alternative approach that benefits from retaining UTRs. We recommend LR Frac-seq for the study of translational control in cases in which complete isoform structures and detection of novel isoforms are desired.
A major implication of LR Frac-seq in the field of translatomics is that its library preparation can be modified to enable direct RNA sequencing after fractionation to detect post-transcriptional modifications that are understood to significantly influence translation. For instance, RNA methylation, specifically N6-methyladenosine (m6A), can alter translation efficiency. Pseudouridylation can affect translation dynamics by influencing ribosome stalling and pausing during protein synthesis. RNA editing events, such as adenosine-to-inosine (A-to-I) editing, can modify regulatory sequences, altering the fate of mRNAs. These post-transcriptional modifications exemplify some of the multifaceted ways in which RNA modifications can impact translational control. By coupling accurate positions of post-transcriptional modifications with polyribosome profiles at isoform resolution, LR Frac-seq could enable more direct correlation of modifications with their effects on translation. Because we used R2C2, which is a cDNA method, to strengthen the confidence of isoform structures, we did not capture modification information beyond RNA editing events. But future adopters of LR Frac-seq can employ direct RNA sequencing methods after fractionation to gain that additional layer of data.
In conclusion, LR Frac-seq enables polyribosome profiling at isoform resolution, retaining complete information about UTRs and novel transcript structures. We tested this method in the context of neuronal differentiation, revealing thousands of transcripts enriched in subcellular fractions relative to the cytosol and largely cell type–specific patterns of isoforms-specific sedimentation between ESCs and NPCs. Our results present intrinsic transcript features and known and novel RBP motifs that may be important determinants of ribosome association, and this work presents a promising new approach to study translational control without the information loss suffered by ribosome profiling and short-read sequencing–based methods.
Methods
H9 cell culture and differentiation to NPCs
H9 cells in feeder-free culture were disaggregated using Accutase and resuspended in hESC medium (StemMACS) containing 10 µM ROCK inhibitor (Y27632). Cells were then seeded on a Matrigel-coated 12-well plate at 50,000 live cells per well. ROCK inhibitor was withdrawn the next day, and the cells were cultured in hESC medium for 3 days. Neural differentiation was then induced over 7 days using KSR medium (for 500.5 mL stock: 415 mL KO-DMEM, 75 mL KSR, 100× Glutamax, 100× NEAA, 1000× bME, 10 µM SB431542, 100 nM LDN-193189). A subset of differentiated cells were stained for PAX6 to confirm neural differentiation.
Short-read Frac-seq
Cytosolic extracts from monolayer-cultured H9 cells and H9-derived NPCs, both in triplicate, were separated on sucrose gradients as described in the original Frac-seq publication (Sterne-Weiler et al. 2013). From these, the Mono fraction (RNAs associated with one ribosome), LPR fraction (two to four ribosomes), and HPR fraction (five or more ribosomes) were isolated using Gradient Station (Biocomp). RNA was extracted with TRIzol, poly(A)-selected, and converted to directional RNA-seq libraries (BIOO Scientific qRNA) from these fractions in addition to total cytosolic RNA. Biological and technical replicates were sequenced using HiSeq 4000 PE150 (50 million to 100 million reads per library).
Long-read Frac-seq
From the same fractionated mRNA used prior for Illumina sequencing, full-length cDNA was prepared using the rolling circle amplification to concatemeric consensus (R2C2) method (Volden et al. 2018). Libraries were pooled and sequenced on an ONT PromethION, generating 12.11 million reads with read length N50 of 17.6 kb.
De novo transcriptome assembly from long reads
R2C2 long reads were base-called with Bonito v0.0.1 (https://github.com/nanoporetech/bonito). Subsequent poly(A) tail and adapter trimming followed by definition of high-confidence isoform consensus sequences was carried out using Mandalorion v4.0.0 (Volden et al. 2023) with all sample FASTAs (from ESCs and NPCs, all subcellular and cytosolic fractions in duplicate) as input. The resultant transcriptome was filtered for redundant transcripts using GFFCompare v0.12.6 (Pertea and Pertea 2020) against the GRCh38.p13 release 41 primary assembly annotation (Harrow et al. 2012) and then further filtered and annotated using SQANTI3 v5.1.1 and IsoAnnot Lite v.2.7.3 (de la Fuente et al. 2020). SQANTI3 filtering was done using the machine learning filter with a training set proportion of 80% and a correct classification probability threshold of 70%. The final, filtered long-read transcriptome was then merged with the GRCh38.p13 release 41 primary assembly annotation (Harrow et al. 2012), producing a “comprehensive transcriptome,” to account for transcripts that were potentially missed by long-read sequencing.
Short-read data analysis
Short reads were adapter-trimmed with cutadapt and then mapped to the GRCh38.p13 primary assembly genome with the comprehensive transcriptome annotation using STAR v2.7.8a (Dobin et al. 2013). Transcript-level quantification was performed from the alignments using Salmon v1.9.0 (Patro et al. 2017) in alignment-based mode. Differential expression analysis at the gene, transcript, and isoform levels was carried out using tappAS v1.0.7 (de la Fuente et al. 2020), which utilizes maSigPro v1.72.0 with the following analysis parameters: polynomial degree of three, significance level of 0.05, R2 cutoff of 0.7, fold change of two, and 9000 clusters. Differential expression analyses were performed for each subcellular fraction against its cognate cytosolic fraction (all in triplicate) for each cell type, as well as between subcellular and cytosolic fractions across ESC and NPC. Pathway analyses were done using Metascape (Zhou et al. 2019).
AS analysis was performed using junctionCounts (Supplemental Codes 4, 5; Ritter et al. 2024; https://github.com/ajw2329/junctionCounts), which identifies and quantifies binary splicing events from RNA-seq data, including alternative 5′ and 3′ splice sites (A5SSs and A3SSs), alternative first and last exons (AFEs and ALEs), SEs, RIs, and mutually exclusive exons (MXEs). cdsInsertion (Supplemental Code 1) and findSwitchEvents (Supplemental Code 3) were used to call NMD events. junctionCounts and its partner utilities are publicly available on GitHub (https://github.com/ajw2329/junctionCounts, https://github.com/ajw2329/cds_insertion), and the versions of the executables used are available as Supplemental Codes 5, 1, respectively. Events with any junction read support were included for dispersion estimates and to determine significance of event differences between conditions, with significant events meeting a requirement of 15 or more total junction read support across fractions for each cell type. AS events were statistically tested by comparing the dispersions of junction support for their included and excluded forms using DEXSeq v1.46.0 (Supplemental Code 2; Anders et al. 2012). Events were considered significant if they had 0.1 ≤ Ψ ≤ 0.9 and adjusted P-value ≤ 0.05 when assessing splicing within a condition or had |ΔΨ| ≥ 0.1 and Q-value ≤ 0.05 when assessing changes in splicing across conditions.
Feature analysis
Transcript features were collected from the transcriptome IsoAnnot Lite annotation and by using custom Python scripts (available as Supplemental Code 6 and at GitHub (https://github.com/ajw2329/cds_insertion), including length measurements of transcript, CDS, uORF, and 5′ and 3′ UTRs. Total counts were of 5′ UTR (TOP and UNR_BS) and 3′ UTR (BRD-BOX, CPE, DMRT1_RE, GY-BOX, K-BOX, MBE, and UNR_BS) motifs, uORFs, and repeat sequences (DNA/hAT-Charlie, DNA/TcMar-Tigger, LINE/L1, LINE/L2, low complexity, LTR/ERVL-MaLR, retroposon/SVA, simple repeat, SINE/Alu, SINE/MIR, and srpRNA). Binary features include coding/noncoding, proximal/distal poly(A) tail usage, predicted NMD/no NMD, and intron retention/no intron retention. And, lastly, codon frequencies and GC-content of the transcript, CDS, and 5′ and 3′ UTRs were included. Feature selection for binary classification between transcripts belonging to subcellular fractions was performed using the RFC method from the sklearn.ensemble module of scikit-learn v1.2.2 (https://scikit-learn.org/stable) and evaluated using permutation importance from the sklearn.inspection module. RFC models were generated with the interest of identifying predictive features of ribosome association and were limited by the relatively small subsets of transcripts classed as associated with a particular subcellular fraction.
Motif analysis was performed using HOMER v4.11 (Heinz et al. 2010). Target sequences were produced by slicing FASEs (in the Mono relative to cytosol, the LPR fraction relative to cytosol, and the HPR relative to the Mono and the LPR separately) into 30 nt windows. Each set was subjected to de novo motif discovery against background sets of 30 nt windows produced from SEs that were not enriched in their given fraction. Significant motifs (P-value ≤ 0.05, FDR ≤ 0.2) plus a set of known RBP motifs—CISBP-RNA H. sapiens (Ray et al. 2013)—were then tested for enrichment across all sets of windows in each fraction using SEA v5.5.4 (Bailey and Grant 2021). Motif enrichment scores were filtered for P-value ≤ 0.05. De novo motifs enriched in at least one set of windows were compared with RBP motifs in the Ray 2013 H. sapiens data set (Ray et al. 2013) for potential matches using Tomtom v5.5.4 (Gupta et al. 2007). The best RBP motif match (P-value ≤ 0.05, Q-value ≤ 0.2) for each de novo motif was assigned accordingly.
The same approach to motif analysis was taken with transcripts exhibiting divergent isoform sedimentation between cell types; 30 nt windows were generated for the CDS, 3′ UTR, and 5′ UTR of each such isoform. De novo motif discovery and enrichment were performed on windows from sets of isoforms preferentially sedimenting with each fraction in each cell type versus those preferentially sedimenting with the same fraction in the other cell type. These isoforms had inverse sedimentation profiles, meaning that those that preferentially sediment with a fraction in ESC show the opposite sedimentation in the same fraction in NPC.
Luciferase reporter assays
Luciferase reporters designed to test translational control by alternative first exon sequences were assembled from gene blocks (IDTDNA) and cloned into pLightSwitch 5′ UTR report (Switchgear Genomics). HEK293 cells, grown on six well plates in DMEM supplemented with 10% FCS, were transfected with 2.5 µg pLightswitch reporter plasmid and pMIR (Ambion). Twenty-four hours after transfection, cells were lysed with passive lysis buffer and analyzed by dual luciferase assay (Promega). Experiments were performed in triplicate. Relative luciferase activity (Renilla vs. firefly) was plotted in Graphpad and analyzed by paired t-test.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE244655.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was made possible by a grant from the California Institute for Regenerative Medicine (GCIR-06673-A). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or any other agency of the State of California. This work was also supported by grants from the National Institutes of Health (National Institute of General Medical Sciences) R35GM130361.
Author contributions: A.J.R. performed data processing and bioinformatic analyses and wrote the paper; J.M.D. designed and performed experiments. J.R.S. conceptualized the project and designed experiments. C.V. helped J.M.D. with performing R2C2 library preparation and data analysis.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279170.124.
-
Freely available online through the Genome Research Open Access option.
- Received February 20, 2024.
- Accepted May 21, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








