
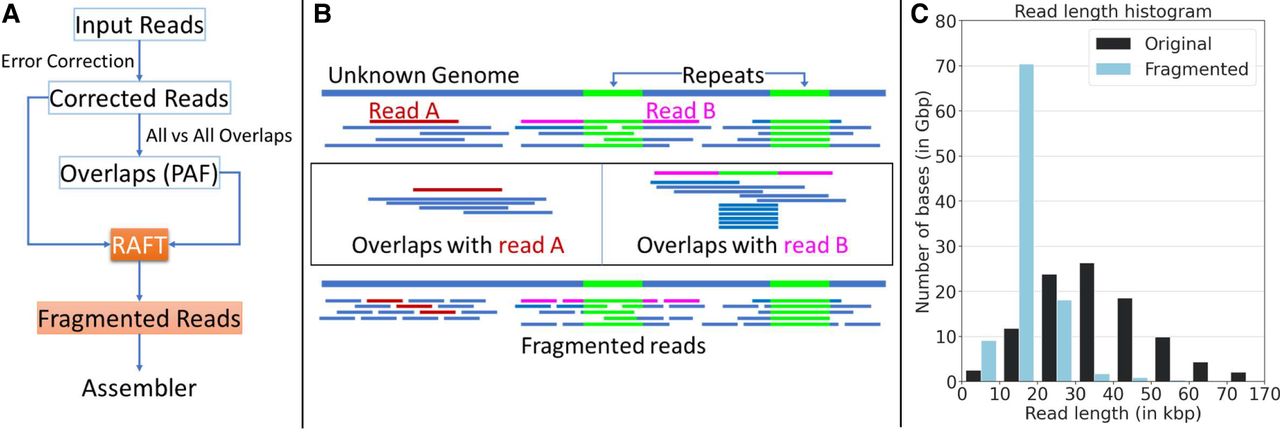
Illustration of the RAFT algorithm and its usage for genome assembly. (A) Flowchart of an assembly workflow that uses RAFT. RAFT accepts error-corrected long reads and all-to-all alignment information as input. It produces a revised set of fragmented reads with a narrow read-length distribution. (B) Illustration of the RAFT algorithm. Read A (shown in red) is sampled from a nonrepetitive region of the genome. Accordingly, RAFT fragments read A into shorter uniform-length reads. Read B (shown in pink) spans a repetitive region of the genome. RAFT detects the repetitive interval in read B because more than the expected number of sequences align to that interval. The portions of read B outside the repetitive interval are split into shorter reads. (C) The impact of RAFT can be seen on a set of ONT Duplex reads sampled from the HG002 human genome. The range of the read lengths is significantly reduced by using RAFT. The original data set comprises 3.7 million reads with a skewed read-length distribution. After fragmentation, the data set comprises 6.8 million reads.








