
Long-read DNA and cDNA sequencing identify cancer-predisposing deep intronic variation in tumor-suppressor genes
- Suleyman Gulsuner1,4,
- Amal AbuRayyan1,4,
- Jessica B. Mandell1,
- Ming K. Lee1,
- Greta V. Bernier2,
- Barbara M. Norquist3,
- Sarah B. Pierce1,
- Mary-Claire King1 and
- Tom Walsh1
- 1Departments of Medicine (Medical Genetics) and Genome Sciences, University of Washington, Seattle, Washington 98195-7720, USA;
- 2UW Medicine–Valley Medical Center, Renton, Washington 98055, USA;
- 3Division of Gynecologic Oncology, University of Washington, Seattle, Washington 98195, USA
-
↵4 These authors contributed equally to this work.
Abstract
The vast majority of deeply intronic genomic variants are benign, but some extremely rare or private deep intronic variants lead to exonification of intronic sequence with abnormal transcriptional consequences. Damaging variants of this class are likely underreported as causes of disease for several reasons: Most clinical DNA and RNA testing does not include full intronic sequences; many of these variants lie in complex repetitive regions that cannot be aligned from short-read whole-genome sequence; and, until recently, consequences of deep intronic variants were not accurately predicted by in silico tools. We evaluated the frequency and consequences of rare deep intronic variants for families severely affected with breast, ovarian, pancreatic, and/or metastatic prostate cancer, but with no causal variant identified by any previous genomic or cDNA-based approach. For 10 tumor-suppressor genes, we used multiplexed adaptive sampling long-read DNA sequencing and cDNA sequencing, based on patient-derived DNA and RNA, to systematically evaluate deep intronic variation. We identified all variants across the full genomic loci of targeted genes, applied the in silico tools SpliceAI and Pangolin to predict variants of functional consequence, and then carried out long-read cDNA sequencing to identify aberrant transcripts. For eight of the 120 (6%) previously unsolved families, rare deep intronic variants in BRCA1, PALB2, and ATM create intronic pseudoexons that are spliced into transcripts, leading to premature truncations. These results suggest that long-read DNA and cDNA sequencing can be integrated into variant discovery, with strategies for accurately characterizing pathogenic variants.
There is now close to consensus that loss-of-function variants in 10 genes—BRCA1, BRCA2, PALB2, ATM, CHEK2, BARD1, BRIP1, RAD51C, RAD51D, and TP53—are responsible for predisposition to breast and ovarian cancer, with other genes responsible for predisposition to breast cancer in special contexts, such as lobular disease (CDH1) or Cowden syndrome (PTEN) (Foulkes 2021). Of these genes, BRCA1, BRCA2, PALB2, RAD51C, and RAD51D are essential for homologous recombination repair of double-strand DNA breaks, and patients with variants in these genes respond well to PARP-inhibitor treatment (Swisher et al. 2021). In addition to cancers of the breast and ovary, individuals heterozygous for inherited loss-of-function variants in these genes are also at increased risk for pancreatic, stomach, and metastatic prostate cancer (Pritchard et al. 2016; Li et al. 2022; Momozawa et al. 2022). It has therefore become best practice in oncology to test for inherited damaging variants in these genes and to profile tumors for somatic mutations in the same genes (Gradishar et al. 2024).
Nonetheless, despite tremendous advances in gene discovery and variant detection, a great deal of the familial risk of these cancers remains unexplained. We hypothesized that one class of not-yet-fully-discovered variation responsible for this increased risk is rare or private deep intronic variants in tumor-suppressor genes that alter splicing and are inherited with cancer in their host families. Long-read genomic and cDNA sequencing, carried out in tandem, are effective in detecting this class of variation. With long-read genomic sequencing, most introns can be sequenced in single reads, thereby disentangling highly repetitive regions, and with long-read cDNA sequencing, single reads span multiple exons, revealing precise splice sites of all transcripts (Glinos et al. 2022). We carried out long-read genomic DNA sequencing and long-read cDNA sequencing for 120 families severely affected with breast, ovarian, or metastatic prostatic cancer, but with no causal variant detected by our BROCA gene panel (Walsh et al. 2010), by exome sequencing or, for a subset, by short-read whole-genome sequencing. For eight of these 120 previously “unsolved families” (6%), a rare deep intronic variant caused exonification of intronic sequence, leading to a stop codon and loss of gene function.
Results
Multiplexed long-read adaptive-sampling genomic sequencing of 240 affected relatives from 120 unsolved families revealed 92 rare or private deep intronic variants in 88 of the families. Each of the 10 targeted genes harbored rare deep intronic variants in at least one family. Targeted genomic regions at the 10 genes and all rare deep intronic variants detected in the 120 families are indicated in the Supplemental Material (Supplemental Tables S1–S3). Analysis by the in silico tools SpliceAI (Jaganathan et al. 2019), the SpliceAI-10k calculator (Canson et al. 2023), and Pangolin (Zeng and Li 2022) yielded scores ≥0.10 for seven of the 92 variants (Supplemental Table S3), suggesting that these variants might create cryptic donor or acceptor sites within introns. These seven candidate variants and, for comparison, 14 of the 85 variants predicted to have no effect on splicing, were evaluated by long-read cDNA sequencing enriched for the candidate gene of each individual (as described in Methods). As the result of targeted enrichment, long-read cDNA sequencing coverage was >100× at the critical gene for each of the samples. All seven variants with scores ≥0.10 yielded transcripts including pseudoexons (Table 1), with all results confirmed by RT-PCR and Sanger sequencing (Supplemental Fig. S1). No pseudoexons were detected by transcript analysis in any of the 14 variants predicted to have no effect on splicing (Supplemental Table S3).
Deep intronic variants leading to pseudoexonification of breast cancer genes
The host families harboring the seven deep intronic variants differed considerably in their cancer profiles.
BRCA1
In families CF3679 and CF6196, BRCA1 c.4987-1352A > G cosegregates with breast, ovarian, pancreatic, and prostate cancer in three generations of two extended families (Fig. 1A). The variant creates a 74 bp pseudoexon in intron 16 with a stop at codon 1673 (Fig. 1B). The families are not closely related, but both trace their ancestry in the 19C to the same region of Sicily. This variant also appeared in two unrelated study participants with ovarian cancer. In family CF4358, BRCA1 c.4358-473T > G is present in the proband with triple-negative breast cancer (TNBC) diagnosed at age 45 and in her sister with ovarian cancer diagnosed at age 49. Their mother and at least one maternal cousin also developed breast cancer (Fig. 2A, left). The variant creates an 84 bp pseudoexon in intron 13 with a stop at codon 1455 (Fig. 2B, left). Family CF4358 is of Mexican ancestry; the same variant was identified in another participant, also of Mexican ancestry, with TNBC diagnosed at age 45. In family CF4455, BRCA1 c.4986 + 69G > A is present in the proband with TNBC diagnosed at age 40. The proband's mother, maternal aunt, and maternal grandmother were diagnosed with ovarian cancer, confirmed by pathology report for the mother and aunt and by death certificate for the grandmother (Fig. 2A, center). The variant leads to extension of exon 16 by exonification of 65 bp of sequence in intron 16, with a stop at codon 1675 (Fig. 2B, center). This variant has not been encountered elsewhere and may be private to this family. For each of the three BRCA1 deep intronic variants, long-read cDNA sequencing with >100× coverage of BRCA1 yielded approximately equal numbers of normal and aberrant transcripts, suggesting that all reads from the variant alleles produced aberrant transcripts.
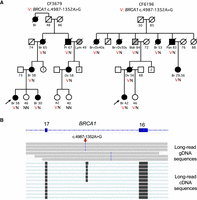
BRCA1 c.4987-1352A > G cosegregates with breast (Br), ovarian (Ov), pancreatic (Pan), and metastatic prostate (Pr) cancer in families CF3679 and CF6196. (A) Filled circles and squares represent females and males with a BRCA1-associated cancer; gray symbols represent persons with other cancers, namely, lymphoma (Lym) and bladder (Bldr). (VN) heterozygosity for the variant, (NN) homozygosity for the normal reference allele. Ages are at diagnosis for cancer patients, at our most recent contact for unaffected living relatives, and at death for relatives who died of a cause other than cancer. (B) IGV images of long-read genomic sequence show the position of the genomic variant (small red arrow) at Chr 17: 43,069,047 (top) and of long-read cDNA sequence including the pseudoexon at Chr 17: 43,069,052–43,069,125 (bottom).
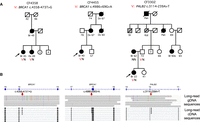
Rare deep intronic variants in BRCA1 in families CF4358 and CF4455 and in PALB2 in family CF3302. (A) Pedigrees have the same notation as in Figure 1, with the addition of cancers of the stomach (St) and kidney (Kid) in family CF3302. (B) IGV images for each of the deep intronic variants show positions of the genomic variants (top) and of long-read cDNA sequences showing the transcriptional profiles created by the genomic variants compared to reference sequences (bottom).
PALB2
In family CF3302, PALB2 c.3114-239A > T is present in the proband with estrogen-receptor-positive breast cancer diagnosed at age 22, which was confirmed as invasive and high grade by pathology. The proband is now age 46, having been diagnosed in intervening years with pituitary and rectal adenomas and in the past year with meningioma in the spine and brain. Her paternal family includes two relatives with pancreatic cancer, which is known to be significantly more frequent among carriers of pathogenic variants in PALB2 (Fig. 2A, right; Casadei et al. 2011). This variant creates a 162 bp pseudoexon with a stop at codon 1058 (Fig. 2B, right). Long-read sequencing of the proband's cDNA with >100× coverage of PALB2 yielded approximately equal numbers of normal and aberrant transcripts, suggesting that all reads from the variant allele produced aberrant transcripts. This variant does not appear in any other family in our studies.
ATM
In family CF5431, ATM c.5763-1080A > G, at Chr 11: 108,309,080 (GRCh38) is present in the proband, diagnosed with clear cell ovarian carcinoma at age 54 (Fig. 3A, left). The variant creates a 112 bp pseudoexon in intron 38 with a stop at codon 1929 (Fig. 3B, left). Long-read cDNA analysis with high coverage of ATM yielded 23% of transcripts with the pseudoexon and 77% normal transcripts, suggesting that the variant allele produced both aberrant and normal transcripts in approximately equal numbers (∼46% vs. ∼54%). This variant has not been previously reported and does not appear in any other participants in our studies, but the ataxia telangiectasia (AT) literature includes reports of AT patients with two other variants located near this position that produce the same pseudoexon (McConville et al. 1996). This cluster of pseudoexonification events may be caused by the distinctive features of this small genomic region. The 35 bp of ATM intron 38 at Chr 11: 108,309,075–108,309,110 is predicted by SpliceAI to be a hotspot for cryptic splicing (Supplemental Fig. S2). This region coincides with an intron of C11orf65 (NM_15287.5), which is transcribed antisense to ATM. In particular, the 35 bp region includes the polypyrimidine tract of the exon 7 splice acceptor of C11orf65. On the ATM strand, this region is AG-rich, with multiple motifs that could be activated as ATM splice donors by 1 bp substitutions. But pseudoexonification also requires a splice acceptor. In this genomic region, a splice acceptor is ready-made at Chr 11: 108,308,956–108,308,968, which are the first base pairs of the 3′ UTR of C11orf65. This short sequence includes two TGA stop codons on the C11orf65 strand, which, together with their flanking base pairs, yield a nearly perfect acceptor motif on the ATM strand (TCATTCATTTCAG). Although this combination of features is unusual, it may not be unique. We speculate that small exons of an antisense gene in introns of a gene on the sense strand may endanger normal splicing of the sense-strand gene.
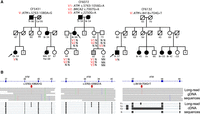
Three rare deep intronic variants in ATM in families CF5431, CF6072, and CF6132. (A) Pedigrees have the same notation as in Figure 1, with the addition of non-Hodgkin's lymphoma (NHL) in family CF5431 and ataxia telangiectasia (AT) in family CF6072. Family CF6072 includes two variants in ATM and one in BRCA2. (B) IGVs for each of the three deep intronic ATM variants showing positions of the genomic variants (top) and of long-read cDNA sequences showing the transcriptional profiles caused by the genomic variants (bottom). ATM c.8418 + 704G > T in family CF6132 yields three different aberrant transcripts, as described in the text.
In family CF6072, ATM c.5763-1056G > A is present in the proband and also in her nephew with AT (Fig. 3A, center). This nephew is compound heterozygous for ATM c.5763-1056G > A and ATM c.2250G > A, a conventional splice variant that leads to transcriptional loss of ATM exon 14. ATM c.5763-1056G > A creates a 137 bp pseudoexon that activates the same cryptic acceptor in intron 38 as the variant in family CF5431, also with a stop at codon 1929 (Fig. 3B, center). Long-read cDNA analysis with high coverage of ATM yielded 30% of transcripts with the pseudoexon and 70% normal transcripts, suggesting that the variant allele generates ∼60% aberrant transcripts. We interpret ATM c.5763-1056G > A as pathogenic but possibly hypomorphic, because ATM c.5763-1050A > G, which also activates the same cryptic acceptor splice site, is well documented as responsible for a relatively mild form of AT (McConville et al. 1996). ATM c.5763-1056G > A clearly contributes to AT in family CF6072. The degree to which it contributes to the breast cancer of the proband is not clear, however, because she is heterozygous both for the ATM intronic variant and for BRCA2 c.7007G > A, a well-documented pathogenic splice variant.
In family CF6132, ATM c.8418 + 704G > T is present in the proband, who was diagnosed with prostatic cancer at age 57 (Fig. 3A, right). The variant creates three aberrant transcripts (Fig. 3B, right). Transcript T1 includes a pseudoexon of 102 bp in intron 57 with a stop at codon 2809 and was 30% of transcripts. T2 includes a pseudoexon of 204 bp in intron 56 with a stop at codon 2762 and was 3.6% of transcripts. T3 includes the pseudoexon of T2 and complete exonification of intron 57, leading to a stop at codon 2762, and was 0.4% of transcripts. Of all ATM transcripts of the proband, 34% were aberrant and 66% were normal, suggesting that the variant allele generates approximately two-thirds (68%) aberrant transcripts and one-third normal transcripts.
Discussion
Many damaging variants responsible for inherited predisposition to cancer alter transcription by introducing or destroying splice sites, leading to premature translation termination. These effects can be caused by variants at or near canonical splice sites, at exonic enhancers, at intronic branchpoints, or at deep intronic sites that activate cryptic acceptor or donor splice sites and introduce pseudoexons (Casadei et al. 2019). For deep intronic variants, evaluation by long-read cDNA sequencing is particularly effective because the long transcripts revealed in single reads enable evaluation of the entire profile of alternate splicing events (Glinos et al. 2022). The goal of this project was to evaluate the combination of long-read DNA sequencing and long-read cDNA sequencing for discovery and characterization of rare damaging deep intronic variants.
This project built on recent observations of deep intronic variants in BRCA1 and BRCA2, as well as on applications of long-read genomic sequencing to detect structural variants in these genes. Based on RT-PCR and Sanger sequencing, BRCA1 c.4185 + 4105C > T was the first deep intronic variant in BRCA1 shown to lead to pseudoexonification and loss of functional protein (Montalban et al. 2019). The likelihood of multiple different variants of this type was suggested by significant enrichment for rare deep intronic variants in BRCA1, BRCA2, and PALB2 among familial breast cancer patients versus cancer-free older women (James et al. 2022). Additionally, a few likely pathogenic deep intronic variants were revealed recently, when Ambry Genetics undertook short-read RNA-seq analysis of thousands of variants that they had classified as VUS, in order to identify those altering splicing (Horton et al. 2024). Nearly all the re-evaluated variants were at or near canonical splice sites, but three were deep intronic variants that Ambry Genetics reclassified as likely pathogenic: BRCA1 c.4185 + 4105C > G (ClinVar accession VCV000632611.7) (see Montalban et al. 2019), BRCA2 c.7618-187G > T (ClinVar accession VCV001759824.2), and BRCA2 c.8332-3384A > T (ClinVar accession VCV001762967.3). A few years ago, in our screen for structural variants, we used long-read sequencing combined with CRISPR-Cas9 excision of BRCA1 and BRCA2 genomic regions to discover an intronic retrotransposon insertion in BRCA1 responsible for inherited young-onset breast cancer in an extended family (Walsh et al. 2021). We subsequently showed that with adaptive-sampling long-read sequencing, the CRISPR-Cas9 excision step could be skipped, eliminating the need for specific CRISPR-Cas9 probes and simplifying library preparation (Miller et al. 2021). Long-read sequencing was also successfully applied to evaluation of pathogenicity of an exonic duplication of BRCA1 (Filser et al. 2023). Genome-wide, long-read cDNA sequencing revealed 70,000 novel transcripts in GTEx tissues and cell lines and demonstrated the feasibility of assessing allele-specific expression mediated by rare cis-regulatory variants (Glinos et al. 2022). Our results suggest that a meaningful number of these rare regulatory variants lie in introns, can be detected in transcripts from blood, and are coinherited with severe phenotypes in families.
Rare deep intronic variants, in genes relevant to a family's phenotype, were encountered frequently. Of the 120 families in this series, 88 families, or 73%, harbored at least one rare or private deep intronic variant in a breast cancer gene. To scale deep intronic screening from 120 families to thousands of families, it is important to prioritize the many genomic variants for cDNA sequencing. Predictions from recently improved in silico tools proved sufficiently precise for this purpose. We found both SpliceAI (Jaganathan et al. 2019; Canson et al. 2023) and Pangolin (Zeng and Li 2022) to be useful, in that all seven variants with at least one score ≥0.10 by either tool, but none of 14 variants with scores near 0.0 by all tools, revealed cryptic splicing. We note that for ATM c.8418 + 704G > T, the highest SpliceAI and Pangolin score was less than 0.20, which the developers of these tools originally considered the threshold for predicting alternate splicing. Because we were interested in this variant, we lowered the threshold for experimental evaluation to 0.10 in order to include it in the study and were rewarded with its complex transcriptional profile (Fig. 3B). Later refinements of the tools suggested that the threshold for follow-up be lowered to 0.05 (Moles-Fernández et al. 2021; Canson et al. 2023). Our parameters enabled us to prioritize experimental evaluation of seven of 92 rare variants (8%), a level of filtering that is compatible with scaling the screen to many unsolved families.
The limitations of long-read sequencing approaches derive both from their inherent features and from challenges in these early days of their development and rapid application. Most critically, long-read genomic sequencing depends on high-quality DNA, and long-read cDNA sequencing depends on high-quality RNA. These requirements will not change; as in the early days of the genome projects, template quality is all-important. It is obviously only possible to obtain sequences of ≥10 kb from DNA of high molecular weight. Availability of high-quality RNA will often be even more problematic, because for some of the most important applications, participants may be very ill. In our experience, the best solution is to generate immortalized lymphoblast cell lines whenever possible from critical participants. Two other constraints of the approach are likely to be resolved soon. Both are computational issues. First, for some indels, SpliceAI currently gives false predictions, which can be artificially either high or low. The SpliceAI lookup site includes an active blog on these problems (https://github.com/broadinstitute/SpliceAI-lookup/issues), so users can determine which classes of indels yield unreliable predictions of splice effects. Second, as long-read-based transcriptome profiles from large numbers of individuals become available, quantification of alternate splice events using tools such as LORALS for cDNA sequencing (Glinos et al. 2022) and NanoCount for direct RNA sequencing (Gleeson et al. 2022) will become more robust.
Finally, the participants in this study are not typical of cancer patients, not even of cancer patients from severely affected families. The participants in this study are members of severely affected families for whom no variant responsible for their cancer had been identified, even after analysis using excellent short-read-based sequencing approaches. The resolution of ∼6% of families from this select “unsolved” group supports the use of long-read sequencing for detection of variants responsible for inherited disease when other approaches have failed to detect a causal gene and allele. For this application, our results suggest straightforward strategies for complete and cost-effective identification of rare deep intronic variants in genomic sequence and for precise characterization of their transcriptional consequences.
Methods
Participants
Participants were individuals with breast, ovarian, pancreatic, or metastatic prostate cancer from families with at least four such relatives (living or deceased) but with no causal variant identified by prior genomic analysis. The number of enrolled affected relatives per family ranged from one to more than 10. For candidate variants, genotypes of deceased affected relatives were reconstructed insofar as possible from informative adult relatives. Complete sequence at all 10 loci was evaluated for 240 affected relatives from 120 unsolved families. The study was approved by the University of Washington Human Subjects Division (study 1583); all participants provided written informed consent.
Genomic sequencing
We used ONT multiplexed long-read adaptive-sampling genomic sequencing (Oxford Nanopore Technologies) to enrich for and sequence 1 Mb regions around the genomic loci of BRCA1, BRCA2, PALB2, ATM, CHEK2, BARD1, BRIP1, RAD51C, RAD51D, and TP53 (for genomic coordinates, hg38 assembly, see Supplemental Table S2). For each participant, 1 μg of high-molecular-weight DNA was sheared to approximately 10–12 kb by two passages through a gTUBE (Covaris) for 1 min at 6000 rpm. Fragments were then end-repaired, A-tailed, and ligated to barcoded adapters (Supplemental Table S1) using the native barcoding kit SQK-NBD114.24. Four barcoded libraries were pooled and 50 fmol loaded onto R10 PromethION flow cells and sequenced for 72 h in adaptive sampling mode enriching for a 10 Mb FASTA reference.
Following sequencing, reads were base-called with the super accuracy model, demultiplexed, and aligned to the target regions, and variants were called with Guppy (Perešíni et al. 2021). Reads were mapped to reference genome (GRCh38) using minmap2 2.26 (Li 2018). BAM files were sorted and indexed using SAMtools 1.15.1 (Danecek et al. 2021). Variants were called by longshot (Edge and Bansal 2019). “Deep intronic variants” were defined as variants >30 bp from a canonical splice site in any of the 10 targeted genes (limiting variants to those >30 bp from splice site excluded variants at branchpoints, which are likely to have different variant profiles). “Rare” variants for BRCA1, BRCA2, PALB2, BARD1, BRIP1, RAD51C, RAD51D, and TP53 were defined as variants with three or fewer entries on gnomAD noncancer v.3.1.2. “Rare” variants for ATM and CHEK2 were defined as variants with 10 or fewer entries on gnomAD noncancer v.3.1.2. Private and rare segregating deep intronic variants were evaluated for splice predictions by SpliceAI (Jaganathan et al. 2019, Canson et al. 2023) and Pangolin (Zeng and Li 2022). On a scale of 0.0 to 1.0, scores ≥0.20 were considered by the creators of these tools to predict possible splice effects, but we evaluated experimentally all variants with scores ≥0.10, as well as an arbitrarily selected subset of variants with scores at or near 0.0.
cDNA sequencing
Total RNA was extracted from blood or lymphoblast cell lines. One microgram of RNA treated with DNase I with a RIN value >9 was reverse-transcribed with a pool of gene-specific primers located in the 3′ UTRs of the 10 target genes (Supplemental Table S4), using the direct cDNA kit (Oxford Nanopore Technologies). We designed and validated gene-specific reverse-transcription primers for each of the 10 breast and ovarian cancer genes. Each oligo (25–30-mer) was specific to the 3′ UTR of its target gene, binds 5′ of poly(A) signal sites, and excludes known SNPs. Individual cDNA libraries (20 fmol) were loaded onto R10 PromethION flow cells and sequenced for 72 h. Following sequencing, reads were base-called with the super accuracy model and aligned to GENCODE transcriptomes in GRCh38 with the splice-aware mode of minimap2 (Li 2018). We defined isoforms using FLAIR v1.4 (Tang et al. 2020) and checked transcripts manually in Integrative Genomics Viewer (IGV) (Robinson et al. 2011). Newly identified exons were validated and their splice junctions confirmed by RT-PCR and Sanger sequencing (Supplemental Fig. S1).
Genotyping
The seven damaging variants were genotyped in anonymized DNA samples from approximately 9000 unrelated individuals with breast or ovarian cancer, using custom TaqMan assays, as previously described (Casadei et al. 2011).
Data access
The long-read sequence data generated in this study have been submitted to the database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) under accession number phs003686.vi.p1.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the families for their continuing enthusiastic participation in our projects. This work was supported by the Breast Cancer Research Foundation (BCRF 23-088), by National Institutes of Health grant 1R35CA197458, and by Department of Defense U.S. Army Research and Development Command grant PC200599. M.-C.K. is an American Cancer Society Professor.
Author contributions: S.G. carried out assembly generation; variant calling, annotation, and merging; data analysis and visualization and wrote major sections of the text. A.A. contributed to cDNA sequencing and analysis; carried out all CLIA validations; and was responsible for curation of cell lines. J.B.M. contacted and counseled families and collected clinical data. M.K.L. contributed to assembly generation and was responsible for overall data management. G.V.B. ascertained and evaluated breast cancer patients. B.M.N. ascertained and evaluated ovarian cancer patients. S.B.P. contributed to transcriptional analysis. M.-C.K. contributed to conceptualization and design of the project, provided project oversight, and wrote major sections of the text. T.W. conceptualized and designed the project, carried out genomic and cDNA sequencing, and wrote major sections of the text. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279158.124.
- Received March 13, 2024.
- Accepted June 20, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








