
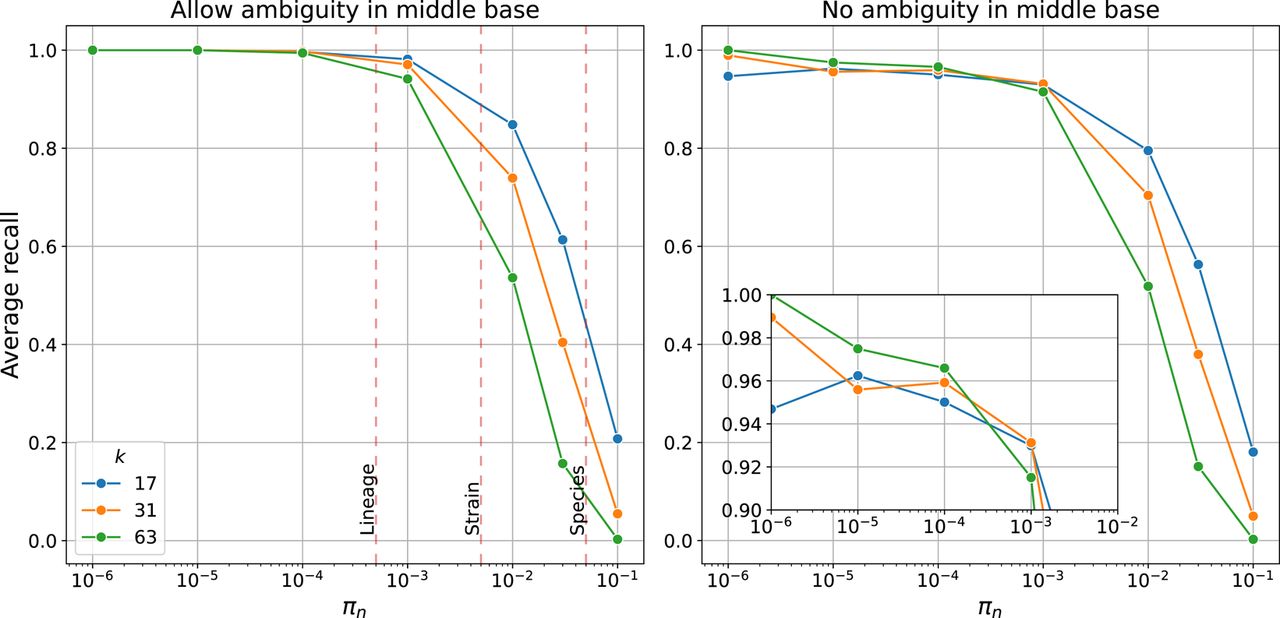
Figure 2.
Average recall of SKA2 in simulations across increasing sequence divergence between a pair of sequences (πn or SNPs per site). Lines show recall using different split k-mer lengths k. (Left) Recall when allowing ambiguous bases, showing typical divergence thresholds used to define species, strain, and lineage boundaries. (Right) Recall when requiring exact matches of the middle base, with inset showing recall over the within-lineage range.








