
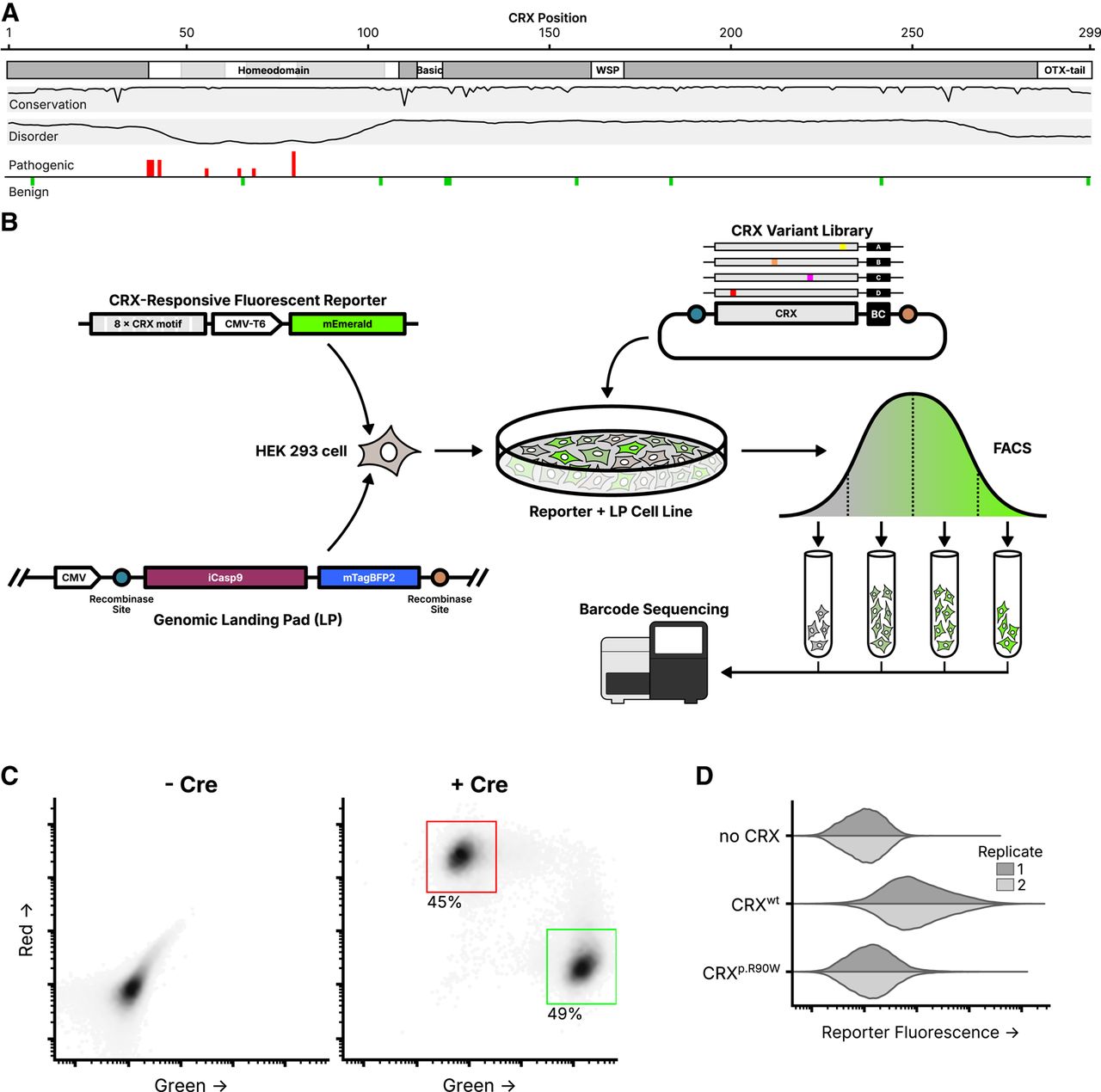
Experimental overview of the CRX deep mutational scan. (A) Known CRX domains, sequence conservation, predicted disorder, and reported ClinVar missense variants. Per-residue conservation was computed using sequences from the UniProt UniRef50 cluster derived from human CRX. Disorder predicted with Metapredict (Emenecker et al. 2021). Missense pathogenic variants (“Pathogenic” and/or “Likely pathogenic”) and benign variants (“Benign” and/or “Likely Benign”) from ClinVar (accessed June 2024); height of bar proportional to number of variants at each position (max = 3). (B) Schematic of the CRX deep mutational scan. A clonal cell line carrying a CRX-responsive fluorescent reporter and genomic landing pad (LP) was generated, and a library of CRX variants was integrated into the LP so that each cell expresses a single variant. Following fluorescence-activated cell sorting (FACS), sequencing was used to determine relative variant barcode abundances in each fluorescence bin, allowing for the calculation of a reporter activity score. (C) LP cells integrated with a 1:1 ratio of plasmids carrying mEmerald (green; arbitrary units) or mCherry2 (red; arbitrary units), with or without a plasmid expressing Cre recombinase (60,000 cells plotted per condition, points shaded by density, percent of cells falling within the indicated gates shown). (D) Reporter activation (green fluorescence; arbitrary units) was measured in Reporter + LP cells with the indicated CRX variants integrated. Two independent biological replicate experiments per sample (distributions plotted from 40,000 cells).








