
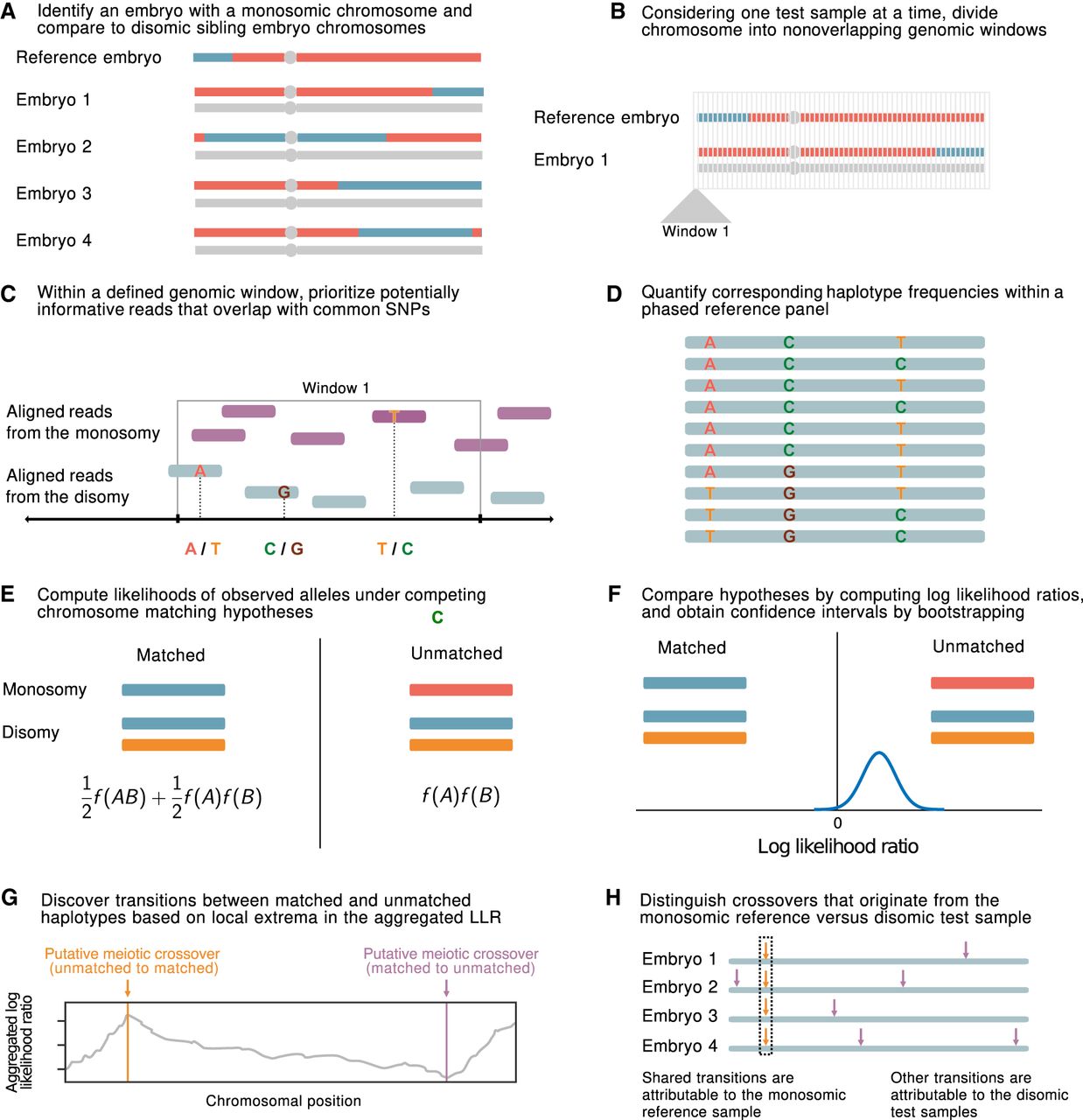
A statistical approach for meiotic crossover discovery based on low-coverage sequencing data from preimplantation genetic testing. (A) Crossover detection is based on haplotype matching between a monosomic chromosome (which is phased by default) and the disomic chromosomes of sibling embryos from the same IVF case. (B) Analysis is conducted within nonoverlapping genomic windows on the scale of 10–100 kbp, defined by the length of typical human haplotypes. (C) Within each window, two to 18 reads are resampled, prioritizing potentially informative reads that overlap common polymorphisms in the population. (D) Frequencies and joint frequencies (i.e., haplotype frequencies) of these SNPs are quantified within an external phased genetic reference panel. (E) Based on these frequencies, the likelihoods of the observed reads are computed under both the matched- and unmatched-haplotype hypotheses. (F) The hypotheses are compared by computing a likelihood ratio, with variance estimated by bootstrapping. (G) Local extrema in the aggregated log-likelihood ratio indicate the locations of meiotic crossovers. (H) Putative crossovers observed in the majority of sibling embryos can be attributed to the monosomic reference chromosome, whereas the remaining crossovers are attributed to the test samples.








