
Histone deacetylases maintain expression of the pluripotent gene network via recruitment of RNA polymerase II to coding and noncoding loci
- Richard D.W. Kelly1,
- Kristy R. Stengel2,
- Aditya Chandru3,
- Lyndsey C. Johnson4,
- Scott W. Hiebert5,6 and
- Shaun M. Cowley1
- 1Department of Molecular and Cell Biology, Henry Wellcome Building, University of Leicester, Leicester LE1 9HN, United Kingdom;
- 2Albert Einstein College of Medicine, Jack and Pearl Resnick Campus, Bronx, New York 10461, USA;
- 3Cancer Research UK Beatson Institute, Bearsden, Glasgow G61 1BD, United Kingdom;
- 4Locate Bio Limited, MediCity, Beeston, Nottingham NG90 6BH, United Kingdom;
- 5Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232, USA;
- 6Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA
Abstract
Histone acetylation is a dynamic modification regulated by the opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs). Deacetylation of histone tails results in chromatin tightening, and therefore, HDACs are generally regarded as transcriptional repressors. Counterintuitively, simultaneous deletion of Hdac1 and Hdac2 in embryonic stem cells (ESCs) reduces expression of the pluripotency-associated transcription factors Pou5f1, Sox2, and Nanog (PSN). By shaping global histone acetylation patterns, HDACs indirectly regulate the activity of acetyl-lysine readers, such as the transcriptional activator BRD4. Here, we use inhibitors of HDACs and BRD4 (LBH589 and JQ1, respectively) in combination with precision nuclear run-on and sequencing (PRO-seq) to examine their roles in defining the ESC transcriptome. Both LBH589 and JQ1 cause a marked reduction in the pluripotent gene network. However, although JQ1 treatment induces widespread transcriptional pausing, HDAC inhibition causes a reduction in both paused and elongating polymerase, suggesting an overall reduction in polymerase recruitment. Using enhancer RNA (eRNA) expression to measure enhancer activity, we find that LBH589-sensitive eRNAs are preferentially associated with superenhancers and PSN binding sites. These findings suggest that HDAC activity is required to maintain pluripotency by regulating the PSN enhancer network via the recruitment of RNA polymerase II.
Acetylation of lysine residues in histone tails is an essential post-translational modification (PTM), dynamically regulated by the opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Verdin and Ott 2015). HDACs are generally considered to be transcriptional repressors (Nagy et al. 1997; Haberland et al. 2009; Kelly and Cowley 2013), because histone deacetylation increases inter-nucleosomal affinity and the association of N-terminal tails with negatively charged DNA, promoting chromatin compaction (Luger and Richmond 1998; Robinson et al. 2008). In addition, loss of histone acetylation prevents the binding of bromodomain-containing proteins, such as TAFII1 (Jacobson et al. 2000) and BRD4 (Kanno et al. 2004), which act as transcriptional coactivators. Among the 18 HDAC enzymes present within mammalian cells, HDAC1, HDAC2, and HDAC3 (class I HDACs) have well-described roles in regulating gene expression as the catalytic core of seven multiprotein complexes: SIN3, NuRD, CoREST, MiDAC, MIER and RERE (HDAC1 and HDAC2), and NCoR (HDAC3) (Millard et al. 2017). Each complex, with its unique arrangements of components, targets HDACs to specific genomic regions through the recognition of histones and interaction with transcription factors (Millard et al. 2017).
Despite a well-established role for HDACs in transcriptional repression, loss of HDAC activity, either through genetic deletion (Zupkovitz et al. 2006; Chen et al. 2013; Dovey et al. 2013; Jamaladdin et al. 2014; Stengel et al. 2017) or through HDAC inhibitors (HDACi) (Karantzali et al. 2008; Baltus et al. 2009; Kim et al. 2013; Greer et al. 2015; Gryder et al. 2019b; Baker et al. 2023), also reduces the expression of a subset of genes. In addition, HDAC activity is required for interferon-induced transcriptional initiation of the immediate-early genes (Chang et al. 2004; Sakamoto et al. 2004). In mammalian cells, HDACs are enriched at transcriptionally active enhancers, promoters, and gene bodies, suggesting an alternative and nonrepressive function (Wang et al. 2009; Kidder and Palmer 2012; Whyte et al. 2012). In tumor cells, HDAC inhibition reduced the activity of superenhancers (SEs) via aberrant hyperacetylation, causing down-regulation of SE-driven transcription of genes such as Myc (Gryder et al. 2019a,b). An influential study from Métivier et al. (2003) showed that activation of the PS2 gene required cyclical acetylation/deacetylation for transcription initiation to occur. In this model, HDACs are hypothesized to function by resetting histone-acetylation at promoters between rounds of initiation (Métivier et al. 2003). In addition to initiation, HDAC activity is also required for normal transcriptional elongation (Kim et al. 2013; Greer et al. 2015). Treatment of cells with HDACi caused increased global histone acetylation levels, resulting in mislocalization of the bromodomain containing 4 (BRD4) (Kim et al. 2013; Greer et al. 2015), an essential cofactor in the conversion of RNA polymerase II (RNAP II) from a paused-to-elongating state (Jang et al. 2005; Yang et al. 2005; Kanno et al. 2014; Arnold et al. 2021). These observations indicate HDACs positively regulate transcription through the precise regulation of histone acetylation patterns.
Embryonic stem cells (ESCs) are widely used to study the transcriptional networks required for pluripotency and tissue-specific differentiation in early embryos (for review, see Martello and Smith 2014). We have previously shown an essential role for HDAC1 and HDAC2 (HDAC1/2) in maintaining the expression of the critical pluripotency-associated transcription factors POU5F1, SOX2 and NANOG (Jamaladdin et al. 2014). Similar transcriptional changes are observed in ESCs treated with HDACi (Karantzali et al. 2008; Baltus et al. 2009) and bromodomain inhibitors (Di Micco et al. 2014; Liu et al. 2014; Horne et al. 2015; Gonzales-Cope et al. 2016; Finley et al. 2018). This is important mechanistically, because bromodomain containing proteins are readers and HDACs erasers of acetylated histones, the latter regulates levels and the former interprets the signal. To further understand the role of dynamic histone acetylation and HDACs in gene regulation, we have examined the molecular mechanisms by which HDACs and the bromodomain and extraterminal motif (BET) family (BRD2-4) maintain expression of the pluripotency-associated gene network.
Results
HDAC1/2 maintain ESC pluripotency in a gene dosage–dependent manner
We previously showed that conditional deletion of either Hdac1 or Hdac2 alone does not alter the expression of pluripotency-associated transcription factors in mouse ESCs (Dovey et al. 2010b). In contrast, double-knockout (DKO) of Hdac1 and Hdac2 caused a reduction in Pou5f1 and Nanog, before a catastrophic loss of cell viability (Jamaladdin et al. 2014). ESCs that retain a single copy of Hdac2 (Hdac1-KO; Hdac2-Het), on the other hand, are viable but have a significant reduction in the total deacetylase activity (Jamaladdin et al. 2014). To analyze the transcriptome in ESCs with differing levels of HDAC1/2 activity, we performed a comparative microarray analysis of DKO and Hdac1-KO; Hdac2-Het cells at 0 (Ctrl) and 3 d after deletion. It takes 4 d for HDAC1/2 to turnover completely, but this time point was chosen to attain the lowest possible amount of HDAC1/2 protein while still retaining cell viability in the DKO cells. Although there were numerous up-regulated transcripts following KO, consistent with a role in gene repression, there were also many genes whose activity was down-regulated by a reduction in HDAC activity. Both DKO and Hdac1-KO; Hdac2-Het ESCs revealed significantly (FDR < 0.05) reduced expression of Pou5f1, Nanog, Esrrb, and Phc1 (Fig. 1A). DKO cells also showed a reduction in additional pluripotency-associated factors, including Klf4, Gdf3, and Sall1 (Fig. 1A, cf. upper and lower panels). Gene set enrichment analysis (GSEA) (Mootha et al. 2003; Subramanian et al. 2005) revealed both DKO and Hdac1-KO; Hdac2-Het cells showed a reduction in pluripotency-associated gene signatures compared with published pluripotent networks (Fig. 1B; Supplemental Fig. S1B). Importantly, reduced expression of pluripotent factors was not accompanied by an increase in differentiation markers (Gata4, Fgf5, or Sox17) (Supplemental Fig. S1A), indicating a direct requirement for deacetylase activity rather than a generalized response to ESC differentiation. To confirm that changes in mRNA were also reflected in protein levels, we examined NANOG and POU5F1 by western blotting. Upon simultaneous deletion of Hdac1/2, we observed a 5.5- and 3.4-fold reduction in NANOG and POU5F1 protein levels, respectively (Fig. 1C). In comparison, compound Hdac1-KO; Hdac2-Het ESCs expressed twofold less NANOG and POU5F1 (Fig. 1C), a smaller reduction than DKO cells, showing a gene-dosage effect of HDAC1/2 activity on pluripotent gene expression. HDAC3 is a highly related homolog of HDAC1/2 that regulates gene expression as a component of the NCoR1/SMRT complex (Watson et al. 2012). We used Hdac3 conditional KO ESCs (Simandi et al. 2016) to address whether reduced expression of pluripotency-associated genes was a general consequence of class I HDAC loss. In contrast to HDAC1/2 DKO cells, cells lacking HDAC3 showed no significant change in Esrrb, Klf4, Nanog, or Pou5f1 (Supplemental Fig. S1C). Taken together, these data show a positive role for HDAC1/2 in the expression of pluripotent factors in ESCs.
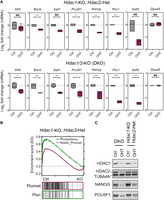
HDAC1 and 2 (HDAC1/2) maintain ESC pluripotency in a gene dosage–dependent manner. (A) Microarray data from n = 3 biological replicates showing relative expression (log2 fold-change) of indicated pluripotency-associated genes in Hdac1/2-KO (DKO) and Hdac1-KO; Hdac2-Het cells at 0 (Ctrl) and 3 d after deletion (OHT). (B) Gene set enrichment analysis (GSEA) plot of microarray data showing Ctrl samples are enriched for the Muller PluriNet (ES = 0.47; P < 0.0001) and pluripotency (ES = 0.70; P < 0.0001) gene sets compared with Hdac1-KO; Hdac2-Het KO cells. (C) Western blot data for HDAC1, HDAC2, NANOG, and POU5F1 proteins in Hdac1/2-KO (DKO) and Hdac1-KO; Hdac2-Het cells at 0 (Ctrl) and 3 d after deletion (OHT). Tubulin, alpha 4A (TUBA4A) was used as a protein loading control.
Acute HDAC inhibition attenuates pluripotency-associated gene expression without inducing differentiation
Investigating the mechanistic relationship between HDAC1/2 and pluripotency using KO models is challenging because the half-lives of HDAC1/2 are ∼24 h, and it takes 3–4 d until the proteins turnover completely (Jamaladdin et al. 2014). Therefore, to evaluate the direct effects of HDAC activity on gene expression in ESCs, we treated E14 cells with the HDACi LBH589 (panobinostat) (Laubach et al. 2015). Because we sought to monitor rapid changes in transcription (at 2–6 h) a hydroxamic acid–based inhibitor was preferable owing to their fast-on/fast-off HDAC-binding kinetics, as opposed to the slow-on/tight-binding kinetics of more specific aminobenzamide inhibitors, for example, MS-275 (Chou et al. 2008; Beconi et al. 2012; Lauffer et al. 2013). To identify HDAC-dependent immediate early genes, we performed a comparative microarray analysis on mRNA isolated from ESCs treated with LBH589 at 2, 6, and 18 h compared with untreated controls. A similar proportion of genes showed increased and decreased expression at all time points, indicating HDAC activity has both positive and negative effects on transcription in ESCs (Fig. 2A). Consistent with the Hdac1/2 DKO model, GSEA showed a reduction in pluripotency-associated gene signatures following treatment with LBH589 (Fig. 2B). Importantly, germ layer differentiation was not stimulated (Fig. 2C), again implying that reduced pluripotency was caused by a reduction in HDAC activity rather than as a consequence of differentiation. We observed reduced expression of Sox2 as early as 2 h, with Esrrb, Nanog, and Klf4 significantly down at 6 h and beyond (Fig. 2D). To confirm that these changes were a direct result of impaired transcription and to remove any confounding effects of mRNA half-life, we pulse-labeled ESCs with 5-ethynyl uridine (EU) to label and purify nascent mRNA, followed by RT–quantitative polymerase chain reaction (qPCR). In the presence of LBH589, nascent Pou5f1 and Nanog transcripts decreased within 1 h of treatment and showed a progressive reduction at 2 and 6 h (Supplemental Fig. S2A). Together, these findings show that acute inhibition of HDAC activity in ESCs directly attenuates pluripotent gene expression without inducing cellular differentiation.
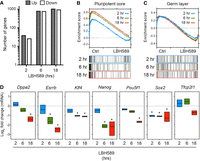
HDAC inhibition attenuates pluripotency-associated gene expression without inducing differentiation. (A) Microarray analysis: the number of genes differentially expressed (1.5-fold; FDR ≤ 0.05) at the indicated times following LBH589 treatment. (B) GSEA plot showing enrichment for the defined pluripotency gene set (Kim_Core_Module) in control versus LBH589-treated ESCs. LBH589 2 h (ES = 0.527; P < 0.0001), 6 h (ES = 0.789; P < 0.0001), and 18 h (ES = 0.83; P < 0.0001). (C) GSEA for Ctrl and LBH589 samples showed equal enrichment for the formation of the primary germ layer (GO:0001704; Germ Layer). (D) Microarray data from n = 3 biological replicates showing relative expression of indicated pluripotency-associated genes in ESCs treated with LBH589 for 2, 6, and 18 h. The statistical difference was calculated using Benjamini and Hochberg's false-discovery rate (FDR). Asterisk denotes significant changes in expression (≤1.5-fold; FDR ≤ 0.05) relative to untreated control ESCs.
Histone acetylation correlates with active transcription in all eukaryotes (Barnes et al. 2019) and is dynamically regulated by HDAC enzymes. We, therefore, addressed the level of H3 Lys27 acetylation (H3K27ac) at the Nanog and Pou5f1 promoters and regulatory regions following HDAC inhibition using chromatin immunoprecipitation (ChIP)–qPCR. The addition of LBH589 stimulated a rapid increase of H3K27ac at cis-regulatory loci, including enhancers of both Nanog and Pou5f1 (Supplemental Fig. S2B), but not at the promoters of developmentally regulated genes (Gata2 and Spink2) in a similar time frame to the reduction in transcription. This is an intriguing result as it shows increased histone acetylation at transcriptionally down-regulated genes, suggesting that active turnover, as part of a transcriptional acetylation/deacetylation cycle (Métivier et al. 2003) or as the global pattern of histone acetylation (Greer et al. 2015), is more important than the simple accumulation of acetylation at a particular locus.
LBH589 and JQ1 target the same subset of pluripotency-associated genes
It has been proposed that increasing global histone acetylation levels using HDACi can sequester the acetyl-lysine reader protein, BRD4, away from critical sites, thereby reducing transcriptional elongation (Greer et al. 2015). BRD4 knockdown (Liu et al. 2014) or inhibition using the bromodomain inhibitor JQ1 (Liu et al. 2014; Horne et al. 2015; Finley et al. 2018) has been reported to reduce the expression of pluripotent-associated factors. To explore the relationship between HDAC activity and BET family members (BRD2-4) in pluripotent gene expression, we treated ESCs with LBH589 (50 nM) and JQ1 (1 µM) for 6 and 18 h. JQ1 treatment reduced the expression of Nanog, Pou5f1, Sox2, and Esrrb to a similar degree and in the same time frame as LBH589, suggesting that there may be a common mode of action (Fig. 3A). We reasoned that if BRD4 levels were limiting Nanog expression, overexpression of BRD4 would rescue the effects of HDAC inhibition. To test this hypothesis, we generated ESCs stably overexpressing BRD4 and, consistent with previous reports (Wu et al. 2015), observed a 4.6-fold increase in Nanog expression (Fig. 3B). However, treatment with LBH589 caused a 2.4-fold (P < 0.0001) and 1.7-fold (P = 0.0241) reduction in Nanog levels with or without additional BRD4, respectively (Fig. 3B). Taken together, these results indicate that sequestration of BRD4 can indeed limit Nanog expression, although the inability of additional BRD4 to rescue the effects of HDACi treatment suggests additional BRD4-independent roles for HDACs at pluripotency-associated genes.
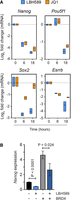
LBH589 and JQ1 target the same subset of pluripotency-associated genes and reduce BRD4 recruitment. (A) RT-qPCR analysis of pluripotency-associated gene expression following the treatment of ESCs with LBH589 or JQ1 for the indicated time points. Box plots represent the max–min expression range and mean from three biological replicates. (B) RT-qPCR analysis for Nanog expression in ESCs treated with LBH589 and/or overexpressing BRD4, as indicated. Values represent mean (±SEM) relative expression compared with the wild-type for n = 6 biological replicates. Statistical differences were calculated using a paired Student's t-test.
HDAC inhibition causes increased expression of promoter upstream transcripts and a loss of promotor-proximal polymerase
To examine the roles of HDACs and BET family members in shaping the ESC transcriptome, we performed precision nuclear run-on and sequencing (PRO-seq) analysis (Kwak et al. 2013). PRO-seq enables the quantification of nascent RNAs and transcriptionally engaged RNA polymerases across the genome, effectively an assay that combines the attributes of nascent RNA-seq with ChIP-seq for RNAP II. To remain consistent with previous experiments, we compared PRO-seq outputs for LBH589-treated cells at 2 h and 6 h and for JQ1 at 2 h. Although our study was primarily focused on the pluripotent gene network, we initially examined the role of HDACs from a global perspective. Analysis of RNAP II occupancies across the transcription start sites (TSSs; ±5 kb) of RefSeq-annotated genes showed significant differences in JQ1- and LBH589-treated cells (Fig. 4A). BRD4 promotes the conversion of proximally paused polymerases to an elongating form via the recruitment of P-TEFb (Jang et al. 2005; Yang et al. 2005). We observed a similar mechanism in our system, as 64% of JQ1-sensitive genes showed an increase in pausing (clusters A, B, and C) (Fig. 4B). Because LBH589 and JQ1 reduced the expression of pluripotency-associated genes (Fig. 3A), our expectation was that loss of deacetylase activity would affect RNAP II pausing in a similar fashion to JQ1. However, in contrast, the majority of genes in LBH589-treated cells showed a reduction in paused polymerase (clusters D, E, and F) (Fig. 4B). These data imply fundamental mechanistic differences in HDAC and BET family function. We also observed increased transcript levels upstream of the TSS in LBH589-treated cells, particularly at genes displaying increased gene body transcription (Fig. 4A,B; clusters C and D). In mammalian cells, active promoters generate short and unstable promoter upstream transcripts (PROMPTs) on the opposite strand to protein-coding genes (Preker et al. 2008). To examine the effects of HDAC inhibition on the generation of PROMPTS, we focused on genes whose expression is up-regulated by LBH589 treatment (clusters C and D). Metagene plots show that LBH589, but not JQ1, produced an increase in PROMPT levels (Fig. 4C). Examination of individual up-regulated genes, Fos and Tpst2 (Fig. 4D), emphasizes the level of increased transcription of PROMPTs following HDAC inhibition. These findings imply HDACs play a role in reducing PROMPT generation, either directly through RNAP II recruitment or indirectly through histone acetylation levels in the vicinity of the TSS.
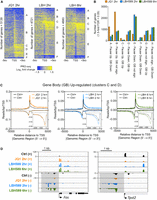
HDAC inhibition causes increased expression of promoter upstream transcripts (PROMPTs) and a loss of promotor-proximal polymerase. (A) Heatmaps displaying a log2-fold change of PRO-seq read counts in 200-bp bins (TSS ± 5 kb) at RefSeq genes displaying increased or decreased pause index changes following JQ1 (2 h) or LBH589 (2 h and 6 h) treatment. Genes were ranked based on the log2-transformed fold-change of RNA polymerase in the promoter-proximal region. (B) A number of gene clusters (A–H) with similar transcriptional changes are shown. For each treatment, genes were clustered based on change (±1.5-fold; FDR ≤ 0.05) in gene body or paused index changes. (C) Metagene plots (TSS ± 5 kb) of RNAP II densities at genes showing increased gene body levels following the treatment with JQ1 or LBH589. Insets show increased antisense transcripts (PROMPTs) relative to TSS in greater detail. (D) PRO-seq Integrative Genomics Viewer (IGV; Robinson et al. 2011) bedGraph screenshots of Fos and Tpst2 TSS with control, JQ1, or LBH589 treatments for the indicated times. Shaded areas indicate increased antisense transcription in LBH589-treated samples.
HDAC activity is required for RNAP II recruitment to pluripotency-associated genes
Pluripotency is the defining phenotype of ESCs. Therefore, among a wide range of differentially expressed genes (4721 at 2 h with LBH589) (Fig. 4A), we chose to examine the pluripotency-associated gene network. We used our PRO-seq data to assess the role of HDACs in transcriptional pausing and elongation among the broader network of factors required for pluripotency. Transcription was reduced in 34 of 59 (2 h) and 45 of 59 (6 h) pluripotent network genes in LBH589-treated cells and 30 of 59 in cells with JQ1 (Fig. 5A), showing a direct requirement for both deacetylase activity and BET family members. Consistent with our initial microarray experiments (Fig. 2A), PRO-seq data showed similar numbers of highly expressed genes are up- and down-regulated with LBH589 (Supplemental Fig. S3), indicating that this is not a generic decrease in abundant transcripts. The reduction in transcription following JQ1 treatment is accompanied by a significant increase in the paused index (PI; the ratio of promoter-proximal to gene body transcript levels) (Fig. 5B; Kwak et al. 2013), indicating that reduced expression may be owing to an inability to resolve paused RNAP II. In contrast, HDAC inhibition produced no consistent change in PI among pluripotency-associated factors (Fig. 5B, cf. JQ1 with LBH589 at 2 and 6 h). Metagene plots of polymerase density among the pluripotent gene network (Fig. 5C) showed increased RNAP II pausing with JQ1, whereas LBH589 treatment caused a reduction in both promoter-proximal and gene body transcription. A reduction in both paused and elongating polymerase can be observed at individual genes such as Nanog, Sox2, and Pou5f1 (Fig. 5D). An overall reduction in transcription suggests that HDAC activity may be required for optimal RNAP II recruitment. To test this further, we performed ChIP-qPCR for RNAP II at the Nanog, Klf4, and Pou5f1 promoters in the presence or absence of LBH589 (Fig. 5E). We observed reduced RNAP II binding at all three genes when treated with LBH589 compared with controls, whereas there was no change with JQ1. Taken together, these findings show that HDAC inhibition reduced the level of both paused and elongating polymerase at pluripotency-associated genes, consistent with a role in RNAP II recruitment that appears to be mechanistically independent of BRD4.
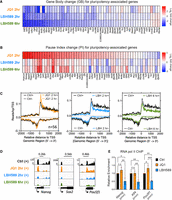
HDACs are required for expression of the pluripotent gene network via recruitment of RNA polymerase II (RNAP II). (A) PRO-seq analysis displaying the effects of JQ1 or LBH589 treatment on the level of gene body transcripts for pluripotent gene network members. Heatmap shows the log2 fold-change relative to untreated controls. (B) Heatmap shows the effects of JQ1 and LBH589 treatment on the pausing index (the ratio of promoter-proximal to gene body transcript levels) changes relative to untreated controls. (C) Metagene plots (TSS ± 2 kb) of pluripotency-associated genes showing reduced initiation and elongation in LBH589-treated samples. (D) RNAP II density at Nanog, Sox2, and Pou5f1 TSS in either control or JQ1- or LBH589-treated ESCs. (E) ChIP-qPCR for RNAP II at the indicated promoters in either control or JQ1- or LBH589-treated (2 h) ESCs. Bars represent the mean (±SEM) of n = 3 biological replicates. (*) P = <0.05, (**) P = <0.01, (***) P = <0.001, (n.s.) not significant.
HDACs positively regulate a subset of enhancers associated with the pluripotent gene network
Analysis of PRO-seq data using nascent RNA sequencing analysis (NRSA) (Wang et al. 2018) identified transcripts in intergenic regions corresponding to enhancer RNAs (eRNAs). Because eRNA levels are a more sensitive indicator of enhancer activity than the histone modifications (Kim et al. 2010), we can directly infer the effects of LBH589 and JQ1 on enhancer activity from their transcription. We observed, consistent with changes in mRNA levels (Fig. 2A), a similar number of down- (199) and up-regulated (149) eRNAs following a 2-h LBH589 treatment, with an increasing number (627 down and 539 up) at 6 h (Fig. 6A). In contrast, the addition of JQ1 predominantly down-regulated eRNAs (1196 down vs. 33 up) and overall affected a far greater number of eRNAs than did LBH589. These data indicate that BET family members play an important role in the expression of eRNAs and that this is independent of HDAC activity.
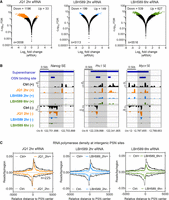
HDACs positively regulate a subset of eRNAs associated with superenhancers (SEs). (A) Volcano plot showing changes in eRNA transcription following treatment with JQ1 or LBH589 for the indicated time points. Statistically enriched (±1.5-fold; FDR ≤ 0.05) eRNA transcripts are indicated in orange, blue, and green. (B) IGV genomic tracks of PRO-seq RNAP II densities at intergenic POU5F1/SOX2/NANOG (PSN) binding sites associated with the indicated SE regions. Shaded areas indicate regions of decreased transcription following LBH589 treatment. (C) Metagene plots of RNAP II density at intergenic PSN binding sites following the indicated treatments.
Both BRD4 (Di Micco et al. 2014) and HDACs (Sanchez et al. 2018) are known to alter transcriptional dynamics at SEs. Therefore, we examined the transcription of eRNAs that overlap with SEs in ESCs treated with either LBH589 or JQ1. A greater proportion of the eRNAs down-regulated by LBH589 overlapped with SEs (39 of 199, 20%) than those with JQ1 (57 of 1199, 5%) (Supplemental Fig. S4). Examination of individual SEs (Nanog, Mycn, and Phc1) shows a distinct reduction in transcription upon HDAC inhibition that appeared unaffected by JQ1 treatment (Fig. 6B, cf. orange and blue/green peaks in shaded regions). Because SEs are defined in part by increased recruitment of lineage-specific transcription factors, we hypothesized that HDAC activity may be required for eRNAs associated with the core pluripotency-associated transcription factors POU5F1, SOX2, and NANOG (PSN). We performed an unbiased analysis of eRNA transcription at all PSN binding sites. Upon HDAC inhibition, we observed a reduction in RNAP II density at both the TSS and the body of the transcript, indicating a reduction in recruitment, whereas JQ1 predominantly affected elongation (Fig. 6C). These data suggest that within 2 h of LBH589 treatment, enhancers associated with PSN function have reduced activity, contributing to the decrease in expression of the entire pluripotent gene network (Fig. 5A).
Discussion
HDAC enzymes and their associated complexes (e.g., SIN3A) are generally viewed as negative regulators of gene expression. The removal of acetyl groups from histone tails leads to a tightening of chromatin that prevents binding of transcription factors. Moreover, transcriptionally silent regions of the genome generally contain hypoacetylated histones (Barnes et al. 2019 and references therein). Despite this, there are several lines of evidence that indicate that HDACs play a role in active transcription. ChIP experiments for class I HDACs indicate that they are recruited to both enhancers and promoters of active genes (Wang et al. 2009; Kidder and Palmer 2012; Whyte et al. 2012). Acetylation/deacetylation cycling may be required to reset promoters between rounds of initiation (Métivier et al. 2003). The latter results have only been shown in a limited number of genes, but the concept is supported by the dynamic turnover of histone acetylation. Acetylation sites within the H2B tail are subject to rapid deacetylation with a half-life of ∼10 min (Weinert et al. 2018). Although these data all support a role in transcription, the readout of histone acetylation remains highly dependent on genomic context, because HDAC inhibition results in both up- and down-regulated genes (Dovey et al. 2010a).
In ESCs, we have shown that HDAC1/2 (Fig. 1A), but not HDAC3 (Supplemental Fig. S1C), are required for expression of the pluripotent gene network. This activity is dependent on gene dosage because a hypomorphic Hdac1-KO; Hdac2-Het cell line has an intermediate level of Pou5f1 and Nanog expression compared with single (Dovey et al. 2010b) or DKO ESCs (Fig. 1A; Jamaladdin et al. 2014). HDAC1/2 are incorporated into six biochemically and functionally distinct multiprotein complexes: SIN3, NuRD, CoREST, MiDAC, MIER, and RERE (Kelly and Cowley 2013; Millard et al. 2017). ESCs in which the NuRD (Kaji et al. 2006; Burgold et al. 2019), CoREST (Foster et al. 2010), and MiDAC (Turnbull et al. 2020) complexes are perturbed still retain pluripotency. In contrast, knockdown of Sin3a (Saunders et al. 2006; Fazzio et al. 2008; Baltus et al. 2009; Adams et al. 2018; Zhu et al. 2018) or SIN3A complex components (Streubel et al. 2017) reduced the expression of Nanog and other pluripotency-associated factors, suggesting that the Sin3 complex may be the critical HDAC1/2 complex component.
Although Hdac1/2 double-knockout ESCs show a robust decrease in Nanog transcription (Fig. 1A), the stable nature of class I HDACs (half-life of 24 h) obliged us to use HDACi to examine the direct effects of deacetylation on transcription. Analysis of nascent transcripts in ESCs showed a significant reduction in Nanog and Pou5f1 transcription within 2 h of LBH589 treatment (Supplemental Fig. S2A). The similarity between LBH589 treatment (a general inhibitor of Zn2+-dependent HDACs) and Hdac1/2 DKO cells suggests that this acute decrease in pluripotency-associated factors is a result of HDAC1/2 inhibition. A critical role of HDAC1/2 in ESCs is to regulate histone acetylation levels (e.g., H3K18ac) across the genome (Kelly et al. 2018), thereby indirectly affecting the recruitment of acetyl-lysine readers, such as BRD4. Increased histone acetylation across the genome could potentially sequester a limiting pool of BRD4 from critical promoters, resulting in a reduced transcription (Greer et al. 2015). In ESCs, we find that overexpression of BRD4 increased the level of Nanog expression, indicating that it is limiting (Fig. 3B). However, overexpression of BRD4 was insufficient to rescue the decrease in Nanog transcription upon HDAC inhibition, arguing in favor of the collaborative but independent roles of BRD4 and HDACs.
To examine the direct roles of HDACs and BET family members (BRD2-4), we performed PRO-seq in ESCs treated with both LBH589 and the bromodomain inhibitor JQ1 (Fig. 4). PRO-seq allows high-resolution mapping of active RNA polymerases across the genome at both coding and noncoding regions and provides information on both the abundance and directionality of transcripts. Analysis of the transcriptome in LBH589-treated ESCs revealed that the majority of genes in LBH589-treated cells showed a reduction in paused polymerase (Fig. 4B, clusters D, E, and F). This contrasts with the addition of JQ1, which produced a significant increase in paused polymerase (Fig. 4B, cluster B), implying fundamental mechanistic differences in HDAC and BET family function. Vaid et al. (2020) performed an analogous PRO-seq study using trichostatin A in Drosophila S2 cells and found that up-regulated genes shared a high degree of pausing, suggesting a release of RNAP II in response to HDAC inhibition. However, it should be noted that our PRO-seq experiments used much longer treatment periods (2 and 6 h) than the Vaid et al. (2020) study (10 and 30 min), resulting in a greater number and variety of dysregulated genes.
RNAP II recruitment to promoters is the principal function of enhancers. Because enhancer activity is associated with the production of eRNAs, whose levels are correlated with the activity of neighboring genes (Andersson et al. 2014; Core et al. 2014), we used our PRO-seq data to examine the contribution of HDACs and BET family members to enhancer function in ESCs (Fig. 6A). JQ1 treatment reduced the transcription of 1196 eRNAs compared with only 199 with LBH589. However, the LBH589-sensitive eRNAs contained a far greater proportion of eRNAs associated with SE activity (Supplemental Fig. S4). The directionality of transcripts identified by PRO-seq also allows the identification of subenhancers contained with SEs. Examination of individual SEs, such as the Nanog, Phc1, and Mycn, shows an overall reduction in transcripts and RNAP II recruitment at each of the subenhancers within these regions (Fig. 6B).
Although our study has identified a role for HDACs in the activity of SEs in ESCs, there is accumulating evidence for their role in the maintenance of SEs in other cell types. Treatment of the human colon tumor cell line HCT116 with the natural HDACi, largazole, resulted in a reduction in RNAP II recruitment to the MYC SE (Sanchez et al. 2018). Moreover, reduced polymerase recruitment was independent of H3K27ac levels, although it remains possible that alternative sites of histone acetylation might be involved (Sanchez et al. 2018). The requirement of HDAC1/2 activity for Myc expression was also confirmed in pancreatic cancer (Stojanovic et al. 2017). In an elegant set of experiments, Gryder et al. (2019b) used constitutive and SE-driven reporters to identify classes of histone-modifying enzymes that preferentially affect SE-driven transcription in alveolar rhabdomyosarcoma cells. They discovered that compared with a variety of bromodomain inhibitors, including JQ1, SEs were more responsive to HDACi (Gryder et al. 2019b). Subsequently, they showed that HDAC inhibition triggers hyperacetylation-induced erosion of SE boundaries, which alters 3D chromatin dynamics and reduces RNAP II occupancy at specific gene loci (Gryder et al. 2019a). In ESCs, we observed an analogous reduction in transcriptionally engaged RNAP II at SEs following LBH589 treatment (Fig. 6B), providing evidence of a broader mechanistic requirement for HDAC activity in different cell types.
Transcription factors that define a cell-specific transcriptional program (such as POU5F1 and NANOG) have increased clusters of binding sites at cell type–specific SEs, which in turn, regulate their own expression (Whyte et al. 2013). This self-reinforcing auto-regulatory network creates a system whereby loss of activity (e.g., treatment with HDACi) leads to a “self-reinforcing” collapse (Gryder et al. 2019b). This hypothesis would fit with our data in ESCs, which showed a reduction in eRNAs associated with PSN binding sites following LBH589 treatment (Fig. 6C). Because POU5F1/SOX/NANOG regulate their own genes, reduction in their protein levels following HDAC inhibition will naturally lead to reduced binding of these factors to their response elements, as well as a general reduction in transcription across the pluripotent gene network (Fig. 5A). In total, we found 4721 differentially expressed genes following 2-h LBH589 treatment (Fig. 4A), indicating that there are additional gene networks to be mined from this data set. Although it has been clear for some time that HDAC inhibition results in both the up- and down-regulation of genes (Clayton et al. 2006; Smith 2008), we have exploited the greatly increased temporal resolution of PRO-seq to begin to unravel the mechanistic basis for these changes in gene expression as a result of perturbing the dynamic regulation of histone acetylation.
Methods
ESC culture and stable BRD4-expressing lines
All mouse E14 ESCs (Hdac1/2L/L, Hdac1L/L; Hdac2L/wt and Hdac3L/L) were cultured as previously described by Jamaladdin et al. (2014). The piggyBAC-BRD4 expression construct was generated by the PROTEX facility at the University of Leicester (https://le.ac.uk/mcb/facilities-and-technologies/protex). The BRD4 cDNA was amplified by PCR, subcloned into pLEICS-38 using the appropriate homology sequences with an in-fusion cloning kit (Takara Bio), and then verified by sequencing (Addgene 83777). The resultant plasmid was transfected into 5 × 105 E14-ESC along with transposase using Lipofectamine 2000. After 24 h after transfection, cells were cultured at low density (5000 cells/10-cm plate) and treated with 200 µg/mL G418 (Thermo Fisher Scientific) in ESC media for 10 d. G418-resistant single colonies were selected, expanded individually, and characterized for FLAG expression by western blotting.
Protein extraction and western blotting
Whole-cell extracts were prepared in cell lysis buffer containing 150 mM NaCl, 50 mM Tris-HCL (pH 7.5), 0.5% IEGPAL, and a protease inhibitor cocktail for 30 min at 4°C. Samples were cleared at 20,000g for 15 min, and the supernatant was used for the examination of soluble proteins. Histones were extracted from the insoluble pellet fraction using 0.4 N sulfuric acid overnight at 4°C. Proteins (30 μg) were resolved using SDS/PAGE and nitrocellulose membranes and were probed with the appropriate antibodies (Supplemental Table S1). The Odyssey infrared imaging system was used to quantify protein signals using the appropriate IR-dye conjugated secondary antibodies (LI-COR Biosciences).
Chromatin immunoprecipitation
ChIP for histone modifications was processed previously as described by Kelly et al. (2018). ChIP for transcription factors was processed by cross-linking cells with 1% formaldehyde for 10 min before being quenched in 125 mM glycine for 5 min. Cross-linked samples were then lysed for 5 min at 4°C in X-ChIP Buffer 1 (50 mM HEPES-KOH at pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, and a protease inhibitor cocktail), before being centrifuged at 900g for 5 min (4°C) and resuspended in X-ChIP buffer 2 (10 mM Tris-HCl at pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, and protease inhibitors) for 5 min at 4°C. Samples were then pelleted (900g for 5 min at 4°C) and resuspended in sonication buffer (10 mM Tris-HCl at pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% EGTA, 0.1% Na-deoxycholate, 0.5% N-laurylsarcosine, 0.1% SDS, and protease inhibitors). Samples were sonicated for 4 min (20 sec on, 40 sec off) using a Bioruptor Pico (Diagenode). After sonication, samples were cleared with 1% Triton X-100 and pelleted (16000g for 5 min at 4°C). Chromatin was immunoprecipitated using 8 μg anti-H3K27ac (Active Motif 39133), 10 μL anti-BRD4 (Bethyl Laboratories A301-985A1), or 10 μL anti-RNAP II (Active Motif 39097) conjugated to 50 µL Dynabeads Protein G overnight at 4°C. A list of the antibodies used can be found in Supplemental Table S1. Beads were washed for 5 min in wash buffer A (50 mM Tris-HCl at pH 7.5, 10 mM EDTA, 75 mM NaCl), wash buffer B (50 mM Tris-HCl at pH 7.5, 10 mM EDTA, 125 mM NaCl), and wash buffer C (50 mM Tris-HCl at pH 7.5, 10 mM EDTA, 175 mM NaCl). DNA was purified using IPure kit v2 (Diagenode) according to the manufacturer's instructions.
RNA extraction and qPCR
Total RNA was isolated using TRIzol (Thermo Fisher Scientific) and Direct-zol RNA Kit (Zymo Research) according to the manufacturer's conditions. Samples were treated with DNase to remove genomic contamination. Five hundred nanograms of RNA was reverse-transcribed using 4 µL Q-Script cDNA SuperMix (Quanta Bioscience) in a 20-µL reaction according to the manufacturer's conditions. cDNA was diluted one in five with DNase- and RNase-free water. For each qPCR reaction, 2 µL of diluted cDNA or ChIP-DNA was amplified in 10-µL reactions containing 2× SensiMix (Bioline) and 1 µM of forward and reverse primers. A complete list of primer pairs, sequences, and annealing temperatures used in the study can be found in Supplemental Table S2. Reactions were performed on a Bio-Rad CFX-Connect under the following conditions: initial denaturation for 10 min at 94°C, followed by 40 cycles for 10 sec at 94°C, primer-specific annealing for 20 sec, and 5 sec at 72°C. Primer efficiency was calculated using absolute quantification from a standard curve. Where applicable statistical differences were identified using a paired two-tailed Student's t-test (Prism v7, GraphPad).
Microarray analysis
Comparative gene expression profiles were generated using the Illumina mouseWG-6, ver.2 (LBH589 and Hdac1L/L; Hdac2L/wt) or Agilent SurePrint G3 Mouse Gene Expression v2 8 × 60 K microarray (Hdac3L/L) according to the manufacturer's instructions. Quality control of total mRNA was performed using a 2100 Bioanalyzer (Agilent), and only RNA integrity numbers (RIN) of eight or higher were selected for processing and array hybridization. LBH589 and Hdac1L/L; Hdac2L/wt arrays were analyzed using Partek Genomics Suite (Partek). GSEA (Subramanian et al. 2005) was performed using GSEA software, version 2.
Precision nuclear run-on assay (PRO-seq)
Nuclear run-on and library construction were performed as previously described (Kwak et al. 2013; Zhao et al. 2016). Briefly, 2 × 107 nuclei were isolated following drug treatments performed in biological replicates. Nuclear run-ons were performed in the presence of 375 µM GTP, ATP, UTP, and biotin-11-CTP with 0.5% sarkosyl for 3 min at 30°C. Total RNA was isolated by TRIzol extraction and hydrolyzed in 0.2 N NaOH, and nascent RNAs were isolated by streptavidin bead binding (Dynabeads MyOne streptavidin T1 magnetic beads, Thermo Fisher Scientific). Following 3′ adapter ligation, 5′ cap removal and triphosphate repair, 5′ hydroxyl repair, and 5′ adapter ligation, RNA was reverse-transcribed, and libraries were amplified. Following PAGE purification, PRO-seq libraries were provided to Vanderbilt Technologies for Advanced Genomics (VANTAGE) for sequencing (Illumina NextSeq 500 SE-75).
Analysis of PRO-seq data, including eRNA analysis, was performed using the NRSA pipeline using alignment files in the BAM format as input (Wang et al. 2018). Adapter trimming was conducted with Trimommatic-0.32 (Bolger et al. 2014) before alignment to mm10 (Mus musculus) genome build using Bowtie 2 (Langmead and Salzberg 2012). All reads <10 bp and ribosomal RNA reads were moved before inputting BAM files in to the NRSA pipeline. Transcriptional rates for genes were determined as follows: Promoter-proximal regions were determining by identifying the largest number of reads in a 50-bp window within ±500 bp from known TSSs; gene body regions were identified within +0.5 kb downstream from a TSS to its transcription termination site (TTS); and pausing indexes were calculated by the ratio of promoter-proximal read density over gene-body read density. For enhancer analysis, NRSA identifies intergenic enhancers as paired bidirectional transcripts having a gap <400 bp between their 5′ ends. Enhancer centers are defined as the midpoints of 5′ end pairs. Transcripts within ±2 kb of any annotated RefSeq gene are excluded from enhancer calling. Down-regulated eRNA overlapping with mouse ESC SE regions were identify using the overlapBed function in BEDTools (Quinlan and Hall 2010). BED files and coordinates for mouse ESC SEs were converted to mm10 genome build using the UCSC liftOver tool.
Metagene plots of nascent RNA levels around regions of interest were generated using the annotatePeaks.pl function in HOMER (http://homer.ucsd.edu/homer/ngs/groseq/groseq.html). POU5F1-SOX2-NANOG (PSN) binding sites were downloaded from and converted to mm10 genome build using the UCSC liftOver tool. Coordinates for TSSs of pluripotency-associated genes, or genes up-regulated following LBH589 treatment, were obtained from UCSC genome (https://genome.ucsc.edu). Average PRO-seq reads around these regions of interest (TSS ± 2 kb or 5 kb) were determined at 25-bp resolution and plotted as histograms in GraphPad Prism version 7 (https://www.graphpad.com/scientific-software/prism/).
SE analysis
Mouse ESC SEs (Whyte et al. 2013) and PSN binding sites (Chen et al. 2008) were converted to the mm10 genome build using the UCSC liftOver tool. BEDTools were then used to identify overlap between SEs and changes in eRNA levels following either LBH589 or JQ1 treatments.
Data access
All raw and processed microarray and sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE136621.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Prof. Ian Eperon, Dr. David English, Dr. Grace Adams, India-May Baker, and Donovan Lim for critical reading of the manuscript. We thank Dr. Nicolas Sylvius and the NUCLEUS Genomics facility for help with performing the microarray data collection; data analysis was assisted by Dr. Matthew Blades. Plasmid constructs were generated by the University of Leicester PROTEX facility. S.M.C. was supported by a senior nonclinical fellowship from the Medical Research Council (MRC; MR/J009202/1) and Biotechnology and Biological Sciences Research Council (BBSRC) project grants (BB/N002954/1, BB/P021689/1). S.W.H. was supported by grants from the Division of Cancer Prevention, National Cancer Institute (NIH; CA164605, CA64140).
Author contributions: R.D.W.K. designed and performed wet-laboratory experiments and conducted bioinformatics analysis. K.R.S. designed and performed wet-laboratory experiments and conducted bioinformatics analysis. A.C. performed bioinformatic analysis of the data. L.C.J. performed functional assays. S.W.H. designed the study and analyzed data. S.M.C. designed the study and wrote the manuscript with input from all coauthors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278050.123.
-
Freely available online through the Genome Research Open Access option.
- Received April 28, 2023.
- Accepted December 20, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








