
Efficient taxa identification using a pangenome index
Abstract
Tools that classify sequencing reads against a database of reference sequences require efficient index data-structures. The
r-index is a compressed full-text index that answers substring presence/absence, count, and locate queries in space proportional
to the amount of distinct sequence in the database: 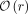 space, where r is the number of Burrows–Wheeler runs. To date, the r-index has lacked the ability to quickly classify matches according to which reference sequences (or sequence groupings, i.e.,
taxa) a match overlaps. We present new algorithms and methods for solving this problem. Specifically, given a collection D
of d documents,
space, where r is the number of Burrows–Wheeler runs. To date, the r-index has lacked the ability to quickly classify matches according to which reference sequences (or sequence groupings, i.e.,
taxa) a match overlaps. We present new algorithms and methods for solving this problem. Specifically, given a collection D
of d documents, 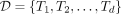 over an alphabet of size σ, we extend the r-index with
over an alphabet of size σ, we extend the r-index with 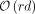 additional words to support document listing queries for a pattern
additional words to support document listing queries for a pattern 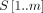 that occurs in
that occurs in 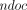 documents in D in
documents in D in 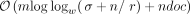 time and
time and 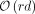 space, where w is the machine word size. Applied in a bacterial mock community experiment, our method is up to three times faster than a
comparable method that uses the standard r-index locate queries. We show that our method classifies both simulated and real nanopore reads at the strain level with
higher accuracy compared with other approaches. Finally, we present strategies for compacting this structure in applications
in which read lengths or match lengths can be bounded.
space, where w is the machine word size. Applied in a bacterial mock community experiment, our method is up to three times faster than a
comparable method that uses the standard r-index locate queries. We show that our method classifies both simulated and real nanopore reads at the strain level with
higher accuracy compared with other approaches. Finally, we present strategies for compacting this structure in applications
in which read lengths or match lengths can be bounded.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277642.123.
-
Freely available online through the Genome Research Open Access option.
- Received January 4, 2023.
- Accepted May 22, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution 4.0 International), as described at http://creativecommons.org/licenses/by/4.0/.








