
Genome skimming with nanopore sequencing precisely determines global and transposon DNA methylation in vertebrates
Abstract
Genome skimming is defined as low-pass sequencing below 0.05× coverage and is typically used for mitochondrial genome recovery and species identification. Long-read nanopore sequencers enable simultaneous reading of both DNA sequence and methylation and can multiplex samples for low-cost genome skimming. Here I present nanopore sequencing as a highly precise platform for global DNA methylation and transposon assessment. At coverage of just 0.001×, or 30 Mb of reads, accuracy is sub-1%. Biological and technical replicates validate high precision. Skimming 40 vertebrate species reveals conserved patterns of global methylation consistent with whole-genome bisulfite sequencing and an average mapping rate >97%. Genome size directly correlates to global DNA methylation, explaining 39% of its variance. Accurate SINE and LINE transposon methylation in both the mouse and primates can be obtained with just 0.0001× coverage, or 3 Mb of reads. Sample multiplexing, field portability, and the low price of this instrument combine to make genome skimming for DNA methylation an accessible method for epigenetic assessment from ecology to epidemiology and for low-resource groups.
Genome skimming refers to unbiased low-pass sequencing below 0.05× coverage and is used to reconstruct mitochondrial genomes as well as identify species and parasites (Twyford and Ness 2017; Papaiakovou et al. 2023; Zhang et al. 2023). Oxford Nanopore Technologies’ nanopore sequencers have been used for genome skimming to produce mitochondrial genomes with success, and at least one study used genome skimming to report DNA methylation (Euskirchen et al. 2017; De Vivo et al. 2022). An important biomarker, DNA methylation varies with age, tissue, species, and environmental exposures, making it a useful measure from ecology to epidemiology (Laubach et al. 2018).
Global DNA methylation measures the ratio of 5′methylcytosines (5 mCs) versus total cytosines reported as a percentage. Current methods to measure global methylation have drawbacks in either cost or accuracy. Antibody-based methods do not have the resolution to distinguish small-magnitude changes often seen in biologically significant epidemiological exposures (Breton et al. 2017). Other methods such as reduced representation bisulfite sequencing (RRBS) and whole-genome bisulfite sequencing (WGBS), although accurate, are expensive, biased in target location, and rely upon bisulfite conversion, which causes downstream bioinformatic challenges.
Nanopore sequencers produce long reads from native genomic DNA, typically in the 10- to 50-kb range, and simultaneously report the presence of DNA methylation genome-wide (Faulk 2022). Long reads enable more contiguous genome assembly and easier reconstruction of repetitive regions (Nurk et al. 2022). Alignment is faster with fewer errors and has the ability to span large transposons and detect structural variants and single-nucleotide variants. Applied to genome skims, long reads enable more accurate recovery of mitochondrial genomes with lower sequencing depth (Cai et al. 2022). Sample multiplexing, field portability, and the low price of the instrument all reduce cost and enable sequencing by low-resource groups (Jahan et al. 2021).
Genome skimming is unbiased in most genomic regions; however, some regions are present in multiple copies, and this enrichment will be reflected in the skimmed read data set. Mitochondria can be present in thousands of copies per nuclear genome and are therefore a common analysis target because a skimming level of 0.1× (300 Mb) would result in hundreds of full-length copies of mitogenomes. Transposons are another overrepresented category in the genome. Repeats make up ∼50% of a mammalian genome on average, with some families present in more than 1 million copies per nuclear genome (Wanner and Faulk 2021). The difficulty in mapping transposons is why WGBS does not provide a true “whole-genome” measure, as its mapping rate is typically ∼50% versus the nanopore mapping rate of >97% we found here. Although few studies have focused on transposons in genome skimming data, the potential is high for transposon biology (Choudhury et al. 2017; Baeza et al. 2021; Sarkar et al. 2023). From an epigenetic perspective, transposons have long been used as proxies for global DNA methylation in epidemiology, so validation of their methylation to global DNA methylation holds promise for their continued use as biomarkers with a lower cost per sample (Faulk et al. 2013).
The aims of this study are (1) to report global DNA methylation levels using genome skimming in the nucleus, in mitochondria, and, specifically, in transposons and (2) to confirm the precision of this method through replication and across species.
Results
Coverage depth estimation
Before designing a genome skimming experiment, it is necessary to determine the minimum level of sequencing depth to obtain sufficient precision for statistical power. A single chimpanzee genome was sequenced to 11.2× depth using a nanopore PromethION instrument and global DNA methylation assessed at 82.87%. Reads were subsampled at coverage levels representing the range of genome skimming expectations, from 0.1× (300 Mb) down to 0.0001× (300 kb), and bootstrapped 10 times to calculate error (Table 1). At 0.01× (30 Mb), coverage of a primate genome results in an error of <1% difference from the true value (Fig. 1).
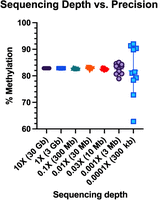
Sequencing depth versus precision. Reads are subsampled from an 11.2× coverage chimpanzee genome at coverage levels from 0.0001× to 10×. DNA methylation is calculated for 10 bootstrapped subsamples.
DNA methylation at varying coverage levels, bootstrapped subsampling
Biological and technical replication
Reproducibility is crucial for assays, which may vary over a small range, and is measured by precision, that is, how close repeated measures are to each other. In the following experiment, mice were sequenced to skimming-level coverage between 0.0025× and 0.016× (median, 0.0058×) or by read coverage, from 8 Mb–133 Mb (median, 47 Mb) reads per sample (Table 2). Three tissues per mouse were sequenced with five mice serving as biological replicates. For controls, whole-genome amplified DNA is expected to have near zero DNA methylation and was used as the low methylated control, whereas CpG methylase–treated DNA is expected to have near 100% methylation and was used as the high methylated control. Multiple aliquots of the same two control reactions were individually barcoded along with the mouse tissues and run on a single MinION flow cell. These served as technical replicates as well as range controls.
Biological and technical replication of DNA methylation in mouse at low coverage
DNA methylation was highly consistent within tissues and significantly higher in testes versus cortex (P < 0.05) (Fig. 2). Quality control validated the experimental protocol (Fig. 3A–D). Methylation values were robust to differences in total bases and average length sequenced, with a fourfold difference in total reads from the lowest group to the highest and a twofold difference in read length between groups. Quality scores did not affect methylation accuracy either, with cortex having a significantly lower, but still high-quality, average q-score between tissues (29.1 vs. 30.2). The lowest q-score occurred in the low methylation control, likely owing to the poorer quality of the in vitro whole-genome amplified DNA. Both controls performed as expected. Technical replication proved very precise with a 0.15% and 0.11% standard deviation for the high and low controls, respectively. All reads reliably mapped to the mouse genome with <0.37% difference across all 24 samples. Mean read quality was slightly lower for the cortex than hippocampus or testes but did not affect precision.
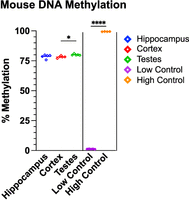
Replicate measures of DNA methylation in the mouse. Hippocampus, cortex, and testes are biological replicates. Control samples are technical replicates. Significance is compared within tissues and controls but not between them. (*) P ≤ 0.05, (****) P ≤ 0.0001.
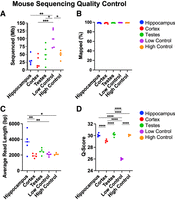
Mouse sequencing quality control. (A) Number bases sequenced in megabases. (B) Percentage of bases mapped to mm39 reference genome. (C) Average read length in base pairs. (D) Average read quality scores. (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001, (****) P ≤ 0.0001.
Vertebrate methylation
Methylation varies with tissue and by species. To determine whether skimming is sufficient to capture primate-specific levels of global DNA methylation, I sequenced buffy coat DNA from five primates to a high depth and down-sampled the reads to a 0.01× equivalent, or 30 Mb (Fig. 4). The global levels are similar to what is seen in human blood.

DNA methylation from primate buffy coat. The bars are colored by genetic distance from human, dark to light.
Across vertebrates, methylation varies widely. Muscle tissue from 34 species was sequenced at a depth ranging from 3 to 200 Mb, median 80 Mb (Fig. 5A). Genome size correlated with percentage of methylation with an R2 of 0.39, likely owing to increased repetitive element content and associated silencing (Fig. 5B). Summary statistics are shown in Table 3; complete data are available in Supplemental File S1.
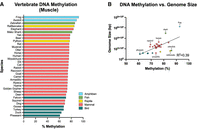
Vertebrate DNA methylation. (A) Global DNA methylation from skeletal muscle in vertebrates. (B) Genome methylation percentage versus genome size in base pairs. Axolotl was removed as an outlier, as it is an order of magnitude larger than other genomes.
Summary statistics for vertebrates by class
Transposons
Genome skimming is unbiased by nature; however, some regions of DNA are enriched compared with single-copy genes; for example, repetitive elements and transposons are present in multiple copies in the nuclear genome and therefore are more likely to be sequenced by skims than single-copy regions. Importantly, DNA methylation plays a strong role in suppressing transposon mobilization and genomic dysregulation. I reasoned that with ∼40% of a typical mammalian genome consisting of repetitive elements, genome skimming can assess methylation at the transposon family level by averaging methylation across all copies of a family.
To determine the minimum depth necessary for precise quantitation, I used the high-depth chimpanzee data set down-sampled from 10× to 0.0001×, quantified DNA methylation, and bootstrapped 10 replicates (Fig. 6; Table 4). For Alu elements, which number more than 1 million copies per cell, even 0.001× coverage, just 3 Mb sequenced, is sufficient for sub-1% accuracy. LINE-1 elements are present in 100,000 copies per cell, although most are truncated. Methylation at LINE-1 at the 0.01× coverage level, 30 Mb sequenced, obtained sub-1% precision. Alu mean methylation was very high at 94.69%, higher than both LINE-1 (90.16%) and the overall genome (82.87%). These values correlate with transposon age and CpG density, where Alus are younger and more CpG dense than LINE-1s, and both are under selection for increased DNA methylation to suppress mobilization.
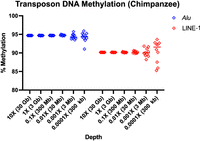
Transposon DNA methylation from chimpanzee buffy coat. From a single 11.72× coverage chimpanzee genome, 10 subsamples of the genome were taken according to coverage level. Alu and LINE-1 methylation are shown for the 10 bootstrap replicates.
Chimpanzee Alu and LINE-1 methylation with 10 bootstrap replicates
The mouse genome skims provide validation and biological replication for transposon methylation assessment. I selected the highest copy number families in mouse, with >1% genomic content threshold, and compared their variability within and across tissues (Fig. 7; Table 5). Above the threshold are SINE families, Alu-B1, B2, and B4; ERV families, class II and ERV-MaLR; and LINE family L1. As in primates, all transposons had higher methylation than the global average of 78.26%–80.07%, depending on tissue. Alu/B1 and L1 elements were stable across tissues, whereas B2s, B4s, and ERVL-MaLRs were significantly increased in testes. ERV class II elements were the only family with a significant decrease in the testes, which is biologically significant as the most frequently active transposon in mice is the intracisternal A particle (IAP), an ERV class II element. These findings have significant implications on transposon based methylation assays.
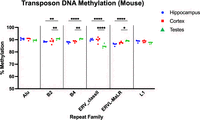
Transposon DNA methylation from mouse. Transposon families above a threshold of 2% genomic content are shown. DNA methylation is calculated from biological replicates of mouse tissues with coverage ranging from 0.0025× to 0.016×. Transposon families with significant differences are shown with ANOVA followed by Tukey's multiple comparison test. (*) P ≤ 0.05, (**) P ≤ 0.01, (****) P ≤ 0.0001.
Mouse repeat DNA methylation by family and tissue
By comparing the methylation of all repeat families across species, patterns emerge (Fig. 8A,B). The mouse repeat complement shows highest methylation in LTRs, SINEs (Alu/B1, B2, B4), ERVs, and LINE-1, which are the most active transposons in the mouse genome. The most prevalent, highest copy number transposons are highlighted in red and tend toward the highest levels of methylation. Similarly in the chimpanzee, SINE Alu and LINE-1 are among the most highly methylated and are the most prevalent in the primate genome. Chimpanzee ERVs remain highly methylated despite having lost mobility in the primate lineage. In both the mouse and chimpanzee, nontransposon repeats have the lowest levels of methylation, for example, low complexity repeats, tRNAs, and simple repeats. Note that the mouse is represented by five replicates taken from hippocampus, whereas the chimpanzee is represented by buffy coat DNA taken from a single individual.
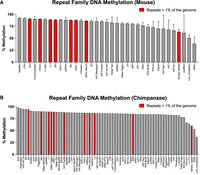
All repeat family DNA methylation. (A) All repeat families from mouse are shown with replication error indicated by error bars. (B) DNA methylation from chimpanzee is shown by repeat family, represented by a single individual. Transposons shown in red make up >1% of the genome of their respective species.
Mitochondria
Genome skimming is frequently used to recover mitochondrial reads for species identification, mitogenome assembly, or other purposes; however, mitochondria are largely unmethylated. To confirm levels seen in other methods, I selected out mitochondria in the mouse samples. I filtered the reads assigned to “chrM” for both mouse tissues and in blood for the chimpanzee. Methylation analysis revealed no significant differences from the low controls (average 0.20%) for each of the three mouse tissues, whereas the high controls averaged 97.34% methylation. The mouse mitochondrial depth ranged from 0.39× to 374× (average 12×) across all tissues, in line with sequencing depth. Because the average mouse sample was sequenced to only 35 Mb total, by increasing sequencing to the 300-Mb level, a coverage of 100× per mitogenome could be generated, enabling efficient assembly of mitogenomes. The mitochondrial-to-nuclear DNA ratio is typically reported as either mitochondria copy number per cell (mtDNA-CN) or as a natural log ratio (Fig. 9). The hippocampus and cortex averaged an mtDNA-CN of 1544 and 1738, respectively. The testes averaged an order of magnitude less at 185 copy number per nuclear genome.
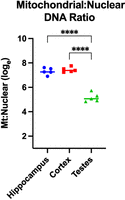
Mitochondrial-to-nuclear (mt:nuc) DNA. The mitochondrial-to-nuclear ratio is significantly reduced in mouse testes. (****) P < 0.0001.
Discussion
Genome skimming, also called low-pass or low-coverage sequencing, is defined as shallow sequencing down to 0.05× coverage of a genome (Li et al. 2021). Here I determined that just 0.01× coverage is sufficient to achieve high precision in global DNA methylation assessment in vertebrate genomes. Genome skimming has been used for diverse purposes, including plastid genome assembly, parasite identification, and evolutionary biology in extinct species, and is gaining software tools for phylogenetic analyses (Grewe et al. 2021; Ferrari et al. 2022; Odago et al. 2022; Reginato 2022; Papaiakovou et al. 2023). Classical genome skimming focuses on mitochondrial genome recovery for species or individual identification but does not typically use the nuclear portion of the skimmed reads or their DNA methylation. There are only six reports of nanopore sequencing used for genome skimming in animals at the time of publication, only one of which examines the epigenome (Euskirchen et al. 2017; Gan et al. 2019; Johri et al. 2019; Franco-Sierra and Díaz-Nieto 2020; De Vivo et al. 2022; Kneubehl et al. 2022). In only one case has genome skimming been used to reconstruct mitochondria with a methylation-aware base caller, but they did not assess either mitogenome or nuclear DNA methylation (Franco-Sierra and Díaz-Nieto 2020). The generation of long reads is crucial to improve alignment over traditional bisulfite-based short reads (Rachtman et al. 2020). The further use of nanopore sequencers for genome skimming will enable the acquisition of DNA methylation in parallel to the DNA sequence data for no additional cost.
Genome skimming for DNA methylation is applicable to any method that generates nanopore sequence data, even in small quantities such as field sequencing. For instance, single-cell skimming is possible because only 30 Mb per sample is needed. With low cost enabled by sample multiplexing, genome skimming becomes ideal for epidemiological exposure monitoring for small-magnitude global changes in DNA methylation in a population. For this experiment, I skimmed 72 samples, yielding 12 Gb of sequencing. A typical minion flow cell can generate 15 Gb of data, so 96 samples could be multiplexed on a single flow cell. Bioinformatic pipelines are maturing, and global methylation is expected to become a commonly reported metric for every type of nanopore run in the future.
To validate the level of precision necessary, I sequenced five biological replicates of three mouse tissues at skimming coverage levels (0.0025× to 0.016×). Detection range and accuracy are confirmed by repeated measures of the technical replicate control samples. Biological replication is validated by highly consistent interindividual measurements among multiple tissues in the mouse biological replicates. Compared with other methods of quantifying DNA methylation, genome skimming is quite accurate and reproducible. Although other methods exist to quantify global DNA methylation, this nanopore-based method has an attractive combination of ease of use, low cost, low barrier to entry, and high precision.
Currently the most common method for global DNA methylation uses a colorimetric antibody-based commercial ELISA kit, which suffers from poor resolution with small-magnitude differences and poor repeatability overall (Cantrell et al. 2020). Further, it only measures the methylated cytosine–to–total cytosine ratio, making its relation to cytosines in CpG context difficult to interpret.
Liquid chromatography uses native DNA degraded to single bases, losing positional information, and suffers from similar challenges as the ELISA method. A recent study using liquid chromatography across the tree of life assessed DNA modifications and reported a large variation in biological replicates. By the nature of the assay, it also reports only the methylated cytosine–to–total cytosine ratio and is difficult to extrapolate to cytosines in CpG context (Varma et al. 2022).
Global methods relying upon the Qiagen Pyromark line of pyrosequencers were formerly popular such as the luminometric assay (LUMA) and the LINE-1, Alu, and mouse IAP amplicon assays (Faulk et al. 2013, 2015). The LUMA assay relies upon isoschizomer methylation-specific enzymatic digestion followed by pyrosequencer quantitation (Karimi et al. 2006). The LINE-1 and Alu assays rely upon bisulfite conversion and determine methylation solely within transposon context, obscuring changes outside transposon regions (Poulin et al. 2018).
I skimmed 40 different vertebrate species across fish, amphibians, reptiles, mammals, and primates, generating global DNA methylation values and transposon methylation. A study using WGBS in multiple vertebrate dermal fibroblasts showed global methylation that recapitulates the pattern of results very well (Al Adhami et al. 2022). The most comprehensive previous study of animal methylation assayed 580 species and matches the global patterns presented here (Klughammer et al. 2023). However, this impressive catalog was created using RRBS and is subject to the typical challenges caused by bisulfite conversion, as well as enrichment for CpG dense regions and lack of accurate transposon identification owing to short reads and loss of mapping ability.
I found that genome size directly correlates to methylation, explaining 39% of the variance, with a clear phylogenetic component. Clade-specific methylation results replicate the phyloepigenetic tree built by Haghani et al. (2021) that was made with a custom 40k epigenetic microarray used on 176 species.
Genome skimming is effective for transposon identification because repeats are present in many thousands of copies per nuclear genome and are known to be methylated at rates near 80% (Crary-Dooley et al. 2017). Transposon methylation in the LINE-1 element is often used as a proxy for global methylation in epidemiological studies (Perera et al. 2019); however, repeat copies are not truly identical and are divided by diagnostic mutations down to the subfamily and individual insertion level. Long reads allow precise identification of repeat type and often identify their exact genomic coordinates. Prior methods to determine DNA methylation of transposons included pyrosequencing, which amplifies a random mixture of transposons of a particular family and quantitates methylation at few specific CpG sites that are of unknown conservation in the bisulfite amplicon, leading to error and poor replication. Even at 0.001× coverage, equating to just 3 Mb of sequence, I was able to accurately determine Alu transposon methylation in the mouse genome, with <1% deviation between biological replicates. Genome skimming has been used in plants to identify and classify the extent of transposon content, but until now, it has not been used to assess DNA methylation of transposons (Choudhury et al. 2017).
I found biologically meaningful results in the mouse transposome, showing that ERV class II elements are substantially hypomethylated in mouse testes compared with other tissues. This is relevant because the most mutagenic transposon in the mouse genome, the IAP element, is an ERV class II, and its hypomethylation in testes suggests germ-line mobilization (Barbot 2002; Faulk et al. 2013).
Genome skimming is often used to sequence mitochondria owing to their naturally high copy number compared with the nuclear genome (Furtwängler et al. 2018). Typical uses are in species identification, assembly of mitogenomes, and ancient DNA haplotyping. In this study, the extraction and library preparation methods are not optimized for mtDNA, and therefore, counts should not be used as absolute values; however, they fall broadly in line with mtDNA: nuclear DNA ratios seen by others (Furtwängler et al. 2018; Longchamps et al. 2020). Even in the tissues with the highest mitochondrial copy number, approaching 1800:1, the mtDNA did not account for >6% of the DNA by weight (or base count), so depletion is not necessary. Intratissue variation in copy number was low, showing consistency and significant differences between tissues. DNA methylation of the mitochondria was extremely low, indistinguishable from the low controls, in line with other studies (Stoccoro and Coppedè 2021).
Regarding nonmodel organisms, good reference genomes are required, which may not exist for a given species. FASTmC is an alternative method for reference-free assessment of methylation using WGBS or inexpensive RRBS (Bewick et al. 2016). Also a new method of enzymatic methyl-seq has become available that mimics bisulfite conversion without the high damage and short reads typical of bisulfite treatment. For bisulfite methods in general, Bismark and other pipelines are well established. Regarding species’ differences in global methylation, other variables beyond genome size could affect methylation, for example, differences in CpG island length and frequency, gene number, and, especially, transposon complement.
Genome skimming with nanopore sequencers is highly effective for global DNA methylation and transposon measurement. Multiplexing along with the ability to pause sequencing allows flexible experimental design and mitigates cost concerns. With improved base calling software and bioinformatic pipelines, global DNA methylation will likely be a basic quality metric for any type of nanopore run. Nanopore sequencing is revitalizing genome skimming in ecology with the detection of cryptic hybridization of threatened primates and in noninvasive methods to assemble mitogenomes (Malukiewicz et al. 2021; Wanner et al. 2021). Extending its utility by adding DNA methylation capability will aid the use of genome skimming in epidemiology, conservation, and comparative genomics.
Methods
DNA extraction
DNA from primate buffy coat samples was extracted Qiagen DNA mini kit (cat. 51306) by the Southwest National Primate Center (SNPRC) and treated with Qiagen RNase A. DNA from mouse tissues and all vertebrate muscle samples was extracted using a Zymo quick-DNA miniprep plus kit (cat. D4068) by the author. Vertebrate skeletal muscle tissues were sourced from commercial meat suppliers or donated by research laboratories. The human muscle sample was obtained commercially from BioChemed Services and was deidentified before purchase. Mice in this study were of the agouti strain, wild-type allele sharing at least 93% similarity with C57BL/6 (Weinhouse et al. 2014). All were males 6–8 wk of age. Mice in this study were maintained in accordance with the guidelines for the care and use of laboratory animals and were treated humanely and with regard for alleviation of suffering. The study protocol was approved by the University of Minnesota institutional animal care and use committee (IACUC). Primate samples were acquired from the SNPRC from incidental blood collection during routine veterinary care and are under the aegis of SNPRC's IACUC approval.
Control DNA was generated by treatment of mouse liver gDNA extracted via a Zymo quick-DNA miniprep plus kit. For the low methylated control, 100 ng of liver gDNA was used as input to a Qiagen REPLI-g mini kit (150023) for whole-genome amplification, following the manufacturer's protocol. The resulting DNA was cleaned using a Axygen AxyPrep MAG PCR clean-up kit (Corning MAG-PCR-CL-50). DNA was incubated with 2× volume of magnetic bead solution on a shaker for 5 min, washed twice with 70% EtOH, and eluted with 100 μL of elution buffer. The yield was 10 μg per reaction. The highly methylated control DNA was generated using 10 μg of liver-derived gDNA as input in separate reactions with a Zymo CpG methylase kit (E2010). Output DNA was cleaned with Axygen beads, and the yield was 70% of input. Control DNA was then used for library prep and sequencing like any other sample extract.
Library preparation and run conditions
DNA from four primates was barcoded and prepped for sequencing using the native barcoding kit SQK-NBD114.24 following the manufacturer's instructions from Oxford Nanopore Technologies. The marmoset sample was prepared with the EXP-NBD104 barcoding kit and the SQK-LSK109 ligation sequencing kit. Primate DNA was sequenced at the University of Wisconsin Biotechnology Center, Sequencing Core on a PromethION 24 instrument. Two samples were multiplexed per PromethION flow cell.
All tissues from mouse and vertebrate muscle were library prepped using the rapid barcoding kit SQK-RBK114.24 following the manufacturer's instructions by the author at the University of Minnesota. Mouse tissue DNA biological replicates along with control DNA were run in a 24-sample multiplex along with technical replicates of five low and four high methylation individually barcoded controls from the same control reaction aliquots on a single MinION R10.4.1 flow cell for 24 h. Vertebrate tissue DNA was run in a 24-sample multiplex on a second MinION R10.4.1 flow cell for 24 h. All MinION runs were performed in the author's laboratory.
Alignment and methylation assessment
The complete bioinformatic pipeline is available in the Supplemental Methods and at GitHub (https://faulk-lab.github.io/skimming/). Briefly, Guppy v6.3.8 was used to call bases for all species with the super accuracy model “dna_r10.4.1_e8.2_400bps_modbases_5mc_cg_sup,” which also calls 5 mC in CpG context. Guppy generates mapped or unmapped BAM files, depending on reference given. Here the reads were aligned post hoc with minimap2 (Li 2018), retaining modification flags by converting the BAM files to FASTQ with SAMtools (Li et al. 2009) v1.16.1 and mapping with minimap2 to create a mapped BAM file. Mapping efficiency was calculated using bamUtils v1.0.16 (https://genome.sph.umich.edu/wiki/BamUtil). Next, the BAM files were converted to BED format with modkit v0.1.4 (https://github.com/nanoporetech/modkit). Summary methylation was calculated with an awk script available in the detailed Supplemental Methods. Read and genome summary statistics were calculated with SeqKit (Shen et al. 2016). Differences in mouse tissue methylation were calculated with an ordinary one-way ANOVA followed by a Tukey's multiple comparison test. Differences in low versus high control methylation were calculated with an unpaired t-test. All mouse sequencing quality tests were compared with ordinary one-way ANOVAs with a Tukey's multiple comparison test.
All software was run on a single computer running Ubuntu v22.04 on an AMD 7950 Ryzen 32 thread CPU with 128-GB memory, 4-TB SSD, and an Nvidia 3080 Ti GPU for accelerated base calling. All statistics and figures were created using GraphPad Prism v9.4.1.
Genomes used in this study are listed in Table 6.
Genome assemblies
Vertebrate statistics
Vertebrate methylation was calculated similarly to that of mouse and primates, with alignment to the reference genome for each species downloaded from NCBI with accession numbers available as Supplemental File S1. Genome size versus methylation excluded axolotl as an outlier because its genome, at 28 Gb, is an order of magnitude larger than any other vertebrate. However, fitting with the pattern, it has the highest methylation percentage of any vertebrate sequenced.
Transposon analysis
Mouse (mm39) and chimpanzee (panTro6) repeatmasked tracks were downloaded from UCSC Genome Browser's Table Browser function with the following parameters: “Table Browser → Repeats → Table “rmsk” → All fields from selected table → repeat-table.tsv.” The table was converted to BED format with a custom awk script. BEDTools was used to intersect the genome-wide methylation BED file generated with modkit versus the repeatmask track BED file. An R script in R v4.2.2 was used to summarize the methylation values by repeat family and statistics applied were applied in GraphPad Prism (R Core Team 2022). Mouse transposon family significance was calculated with a two-way ANOVA followed by a Tukey's multiple comparison test. All scripts are available in Supplemental Methods.
Mitochondria analysis
Mouse and chimpanzee BED files were filtered for CpG sites mapped to “chrM” in each species and assessed for global methylation.
Data access
All processed sequencing data generated in this study are stored as BAM files containing modified base calls and have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA927034.
Competing interest statement
The author declares no competing interests.
Acknowledgments
I thank Arthur Rand, developer of Modkit, for development and updates to improve the accuracy of DNA methylation calls in BAM-to-BED file conversion. This work was supported by National Institutes of Health Grant R21AG071908 (C.F.), Impetus Grant (Norn Foundation; C.F.), and U.S. Department of Agriculture–National Institute of Food and Agriculture Grant USDA-NIFA MIN-16-129 (C.F.).
Author contributions: C.F. conceived the study, performed the sequencing, analyzed the data, and wrote the manuscript. C.F. read and approved the manuscript before submission.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277743.123.
- Received January 27, 2023.
- Accepted June 7, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








