
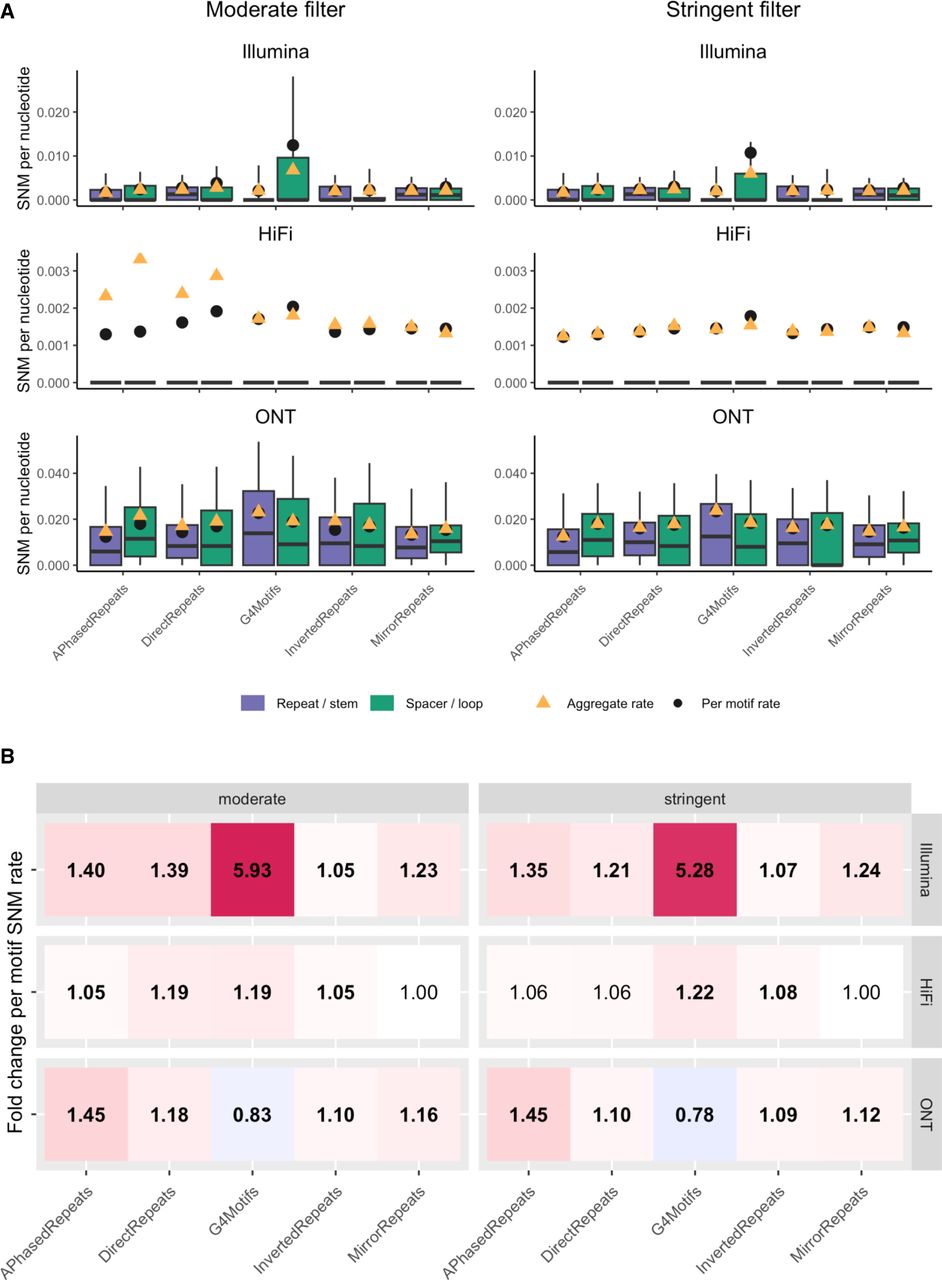
Single-nucleotide error rates in non-B motif subregions. (A) Boxplots of per-motif SNM error rates of subregions of non-B motifs. A-phased, direct, inverted, and mirror repeats are divided into repeat arms and spacer; G4 motifs are divided into stem (G-tract) and loop. Boxplot whiskers show the fifth and the 90th percentiles, and values outside whiskers are excluded from the boxplots in order to better visualize the bulk of the distributions. The left panel shows the moderately filtered set; the right panel, the stringently filtered set. The three rows correspond to the different technologies (Illumina, HiFi, and ONT). Purple and green boxes correspond to repeat/stem and spacer/loop subregions, respectively; black dots mark values for per-motif means; and orange triangles aggregate error rates (sum of all errors divided by sum of all aligned nucleotides). Note that the y-axes differ among technologies. (B) Heat maps visualizing fold changes in per-motif means of SNM error rates between different subregions of non-B motifs. Red (blue) shades indicate higher (lower) error rates in loops and spacers than in stems and repeat arms, with fold change values reported in each cell of the map. When these values are in bold, per-motif means were significantly different between motifs and controls (t-test P-values corrected for multiple testing smaller or equal to 0.05). Also here, left and right panels correspond to moderately and stringently filtered sets, respectively, and rows correspond to Illumina, HiFi, and ONT technologies, respectively.








