
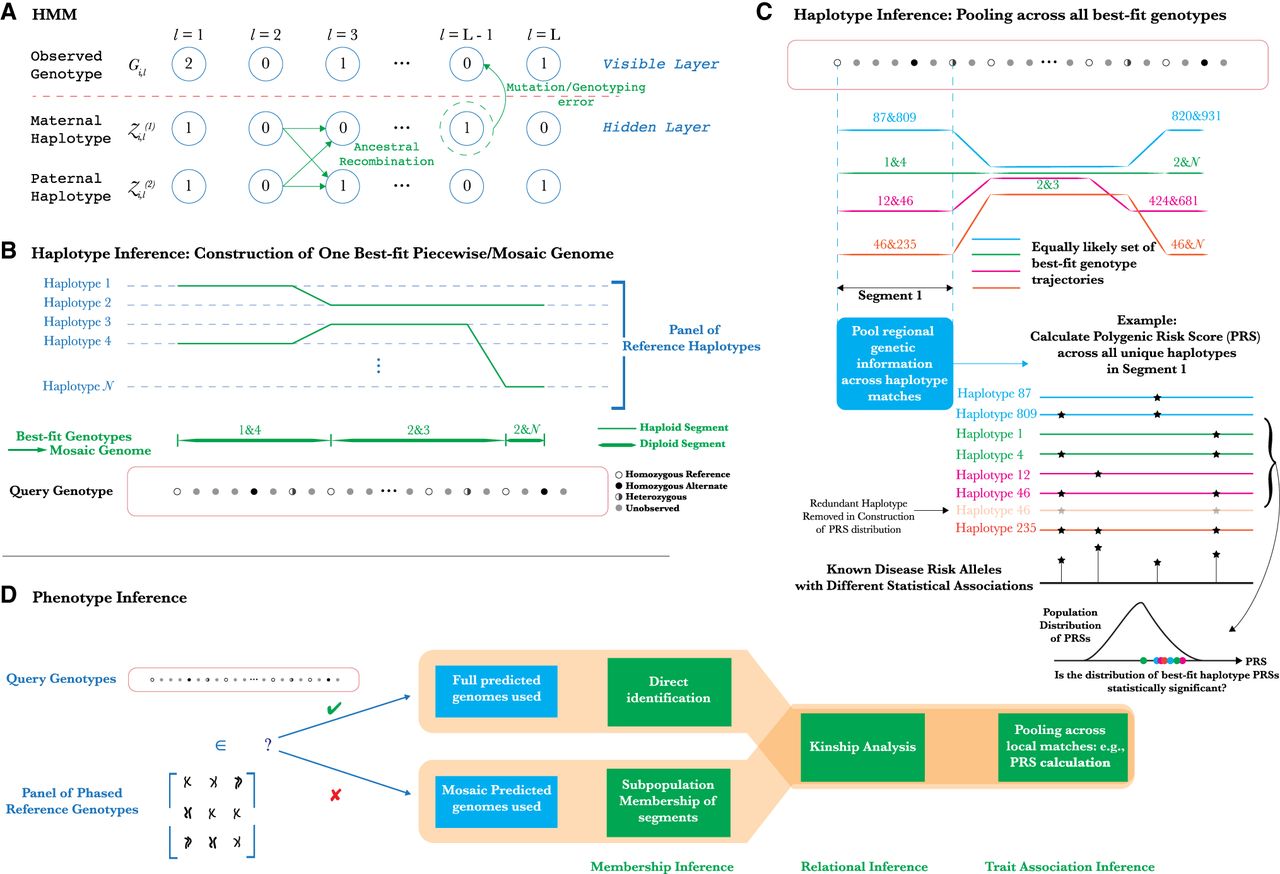
Methodology of PLIGHT's HMM-based inference and downstream analyses. (A) Schematic of the Li-Stephens HMM for haplotype recombination and mutation. (B) Inference of best-fit haplotype pairs in different genomic regions based on a chosen database of reference haplotypes, and the subsequent construction of the diploid mosaic genome. (C) An example of how the haplotype inference procedure described here could be used for novel attacks. By calculating polygenic risk scores (PRSs) for traits across all identified mosaic genome segments, we can test for statistically significant clustering of the PRSs relative to background values. If significant, the individual is more likely to be linked to such a trait. (D) Exploration of potential phenotypic inference attacks, conditional on whether or not the individual is known to be in the reference haplotype database.








