
Abstract
We report the generation of an organism-wide catalog of 976,813 cis-acting regulatory elements for the bovine detected by the assay for transposase accessible chromatin using sequencing (ATAC-seq). We regroup these regulatory elements in 16 components by nonnegative matrix factorization. Correlation between the genome-wide density of peaks and transcription start sites, correlation between peak accessibility and expression of neighboring genes, and enrichment in transcription factor binding motifs support their regulatory potential. Using a previously established catalog of 12,736,643 variants, we show that the proportion of single-nucleotide polymorphisms mapping to ATAC-seq peaks is higher than expected and that this is owing to an approximately 1.3-fold higher mutation rate within peaks. Their site frequency spectrum indicates that variants in ATAC-seq peaks are subject to purifying selection. We generate eQTL data sets for liver and blood and show that variants that drive eQTL fall into liver- and blood-specific ATAC-seq peaks more often than expected by chance. We combine ATAC-seq and eQTL data to estimate that the proportion of regulatory variants mapping to ATAC-seq peaks is approximately one in three and that the proportion of variants mapping to ATAC-seq peaks that are regulatory is approximately one in 25. We discuss the implication of these findings on the utility of ATAC-seq information to improve the accuracy of genomic selection.
Genomic selection has had a tremendous impact on livestock breeding in the past 10 yr (e.g., García-Ruiz et al. 2016). Nevertheless, the accuracy of selection remains inferior to what may be achievable given the heritability of the selected traits. This could have a number of causes, including the size and composition of the reference population or the contribution of dominance and epistasis to the genetic architecture of the traits of interest. Another factor is that all variants are generally given an equivalent weight in computing the additive relationship between animals needed for GBLUP analyses or equivalent prior probabilities of variant effects in Bayesian approaches. Yet, only a minority of variants are causative (having a direct effect on gene function and hence phenotype), with the remainder being, at best, passenger variants in linkage disequilibrium (LD) with one or more of the causative variants. The extent of LD between causative and passenger variants is bound to be population specific, or even subpopulation specific, and is likely to fluctuate over time, and this may account in part for the observed limits in selection accuracy. It is generally believed that knowing the causative variants, or at least those that are more likely to be, may help to further improve the accuracy of genomic selection (Xiang et al. 2019).
Causative variants encompass coding and regulatory variants. Coding variants, including missense, nonsense, frameshift, splice site variants, and deletions, are easily recognized yet only account for a limited part of the genetic variance for complex phenotypes, including production traits. It is increasingly apparent that most of the genetic variation for complex traits is owing to regulatory variants that act either by perturbing the expression profile of genes located in cis (standard polygenic model) or, possibly, by perturbing the gene regulatory network and affecting the expression profile of a restricted number of core genes in trans (omnigenic model) (Liu et al. 2019). Regulatory variants are more difficult to identify as the effect of polymorphisms on the functionality of proximal and distant cis-acting regulatory elements remains difficult to predict. However, it is reasonable to assume that most regulatory variants are located within or in close proximity to regulatory elements, which account for an estimated ∼5%–20% of genome space (Meuleman et al. 2020; The ENCODE Project Consortium et al. 2020). Active regulatory elements can be recognized by virtue of evolutionary constraint (Lindblad-Toh et al. 2011) and epigenetic features, including chromatin accessibility, specific histone codes, transcriptional activity, their participation in loop structures, and transcription factor (TF) occupancy (Meuleman et al. 2020; The ENCODE Project Consortium et al. 2020).
In an effort to identify putative regulatory variants in the bovine, we herein report (1) the generation of a comprehensive catalog of bovine regulatory elements identified using assay for transposase accessible chromatin using sequencing (ATAC-seq) (Buenrostro et al. 2013) in 63 tissue types; (2) the generation of a catalog of common bovine variants that map to identified proximal and distal regulatory elements; (3) the demonstration that variants driving expression quantitative trait loci (eQTL) in liver and blood are more likely to map to regulatory elements that are active in the cognate tissues and, hence, that variants in these regulatory elements are more likely to be causative; (4) estimates of the proportion of regulatory variants that map to ATAC-seq peaks as well as the proportion of variants mapping to ATAC-seq peaks that are regulatory; and (5) a retrospective evaluation of the utility of this catalog for the identification of regulatory variants known in livestock species.
Results
Generating a catalog of bovine cis-acting gene regulatory elements
To generate a bovine catalog of open chromatin regions using ATAC-seq, we collected 106 samples corresponding to 68 tissue types (Fig. 1A; Supplemental Tables S1, S2). Most samples (73%) were collected from the same juvenile Holstein male. The remainder (27% including gonads and mammary gland) were collected from nine additional animals (Supplemental Tables S1, S2). Fresh and frozen samples were subjected to ATAC-seq using standard procedures with two concentrations of tagmentation enzyme (Buenrostro et al. 2013; Corces et al. 2017). We sequenced a total of 185 libraries to an average of 31.8 million paired-end reads per library (Supplemental Table S3). To these in-house-generated data, we added publicly available ATAC-seq data (15 data sets) from five additional tissues/cell types (Fang et al. 2019; Halstead et al. 2020a,b; Johnston et al. 2021). Reads were mapped to the bovine genome (ARS-UCD1.2) with Bowtie 2 (Langmead and Salzberg 2012) and ATAC-seq peaks called with MACS2 (Zhang et al. 2008) following ENCODE's recommendations (https://www.encodeproject.org/atac-seq/). Data sets passing quality control (89/106 in-house-generated data, i.e., 84%) and corresponding to technical replicates were merged (per biosample), resulting in a total of 104 ATAC-seq data sets (89 in-house and 15 public) representing 63 tissue types (58 in-house and five public). Pearson's correlations between technical and biological replicates (normalized read counts across 500-bp windows covering the entire genome) exceeded 0.89 and 0.85, respectively (Supplemental Figs. S1, S2).
Generation of an organism-wide catalog of cis-acting regulatory elements for the bovine. (A) Sixty-three tissue types with ATAC-seq data analyzed in this work. Novel ATAC-seq data were generated for 58 tissue types (89 samples), and public ATAC-seq data were downloaded for five (15 samples). Tissue types are grouped and colored based on the nonnegative matrix factorization (NMF) analysis described in D. Tissues are parenthesized when the largest NMF component in the tissue explains <50% of the total weight. This figure was created with BioRender (https://www.biorender.com). (B) Size distribution of proximal (green) and distal (red) ATAC-seq peaks (consensus peaks). (C) Distribution of the number of samples in which proximal (green) and distal (red) ATAC-seq peaks are open. (D) Distribution of the accessibility (fold-increase in coverage over background) of proximal (green) and distal (red) ATAC-seq peaks. The vertical dotted lines in B, C, and D correspond to the medians. (E) Distribution of GERP scores for nucleotide positions within proximal (solid green) and distal (solid red) ATAC-seq peaks, within sequence segments of same size immediately flanking proximal (dotted green) and distal (dotted red) ATAC-seq peaks, and across the entire genome (gray). The proportion of nucleotide positions without GERP score is not shown. (F) Decomposition of the 976,813-peak × 104-sample matrix in 16 components by nonnegative matrix factorization (NMF) following the method of Meuleman et al. (2020). As a result, each peak and each tissue sample are represented as a linear combination of the 16 components, which are color-coded in the graph. The lengths/heights of the bars measure the loading factor of the corresponding component for each of the tissue samples/peaks. Anatomically related samples typically have the same dominant component and have been ordered accordingly (Supplemental Table S5). The peaks that are predominantly active in the cognate tissue samples are dominated by the same component and are ordered accordingly. Thirty-one samples did not show clear tissue-specific peaks; their ATAC-seq profiles were dominated by the “ubiquitous” peaks shared by nearly all samples and, to a lesser extent, by a group of peaks assigned to the 16th “undefined” NMF component (shown in gray).
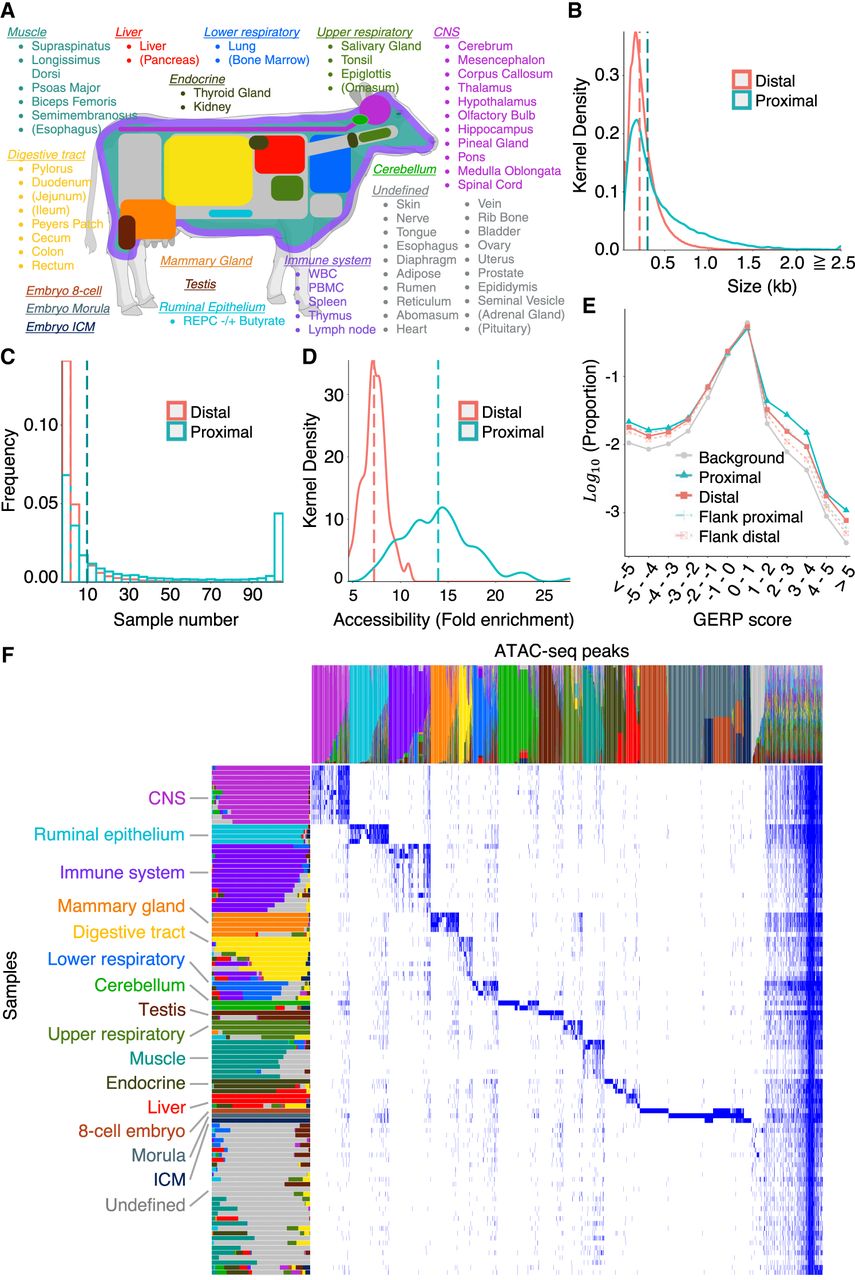
MACS2 yielded an average of 76,919 peaks per sample in ATAC-seq mode (range: 15,420–238,210 peaks) and 51,838 peaks per sample in ChIP-seq mode (range: 14,594–201,757 peaks) (Supplemental Fig. S3; Supplemental Table S4). We merged ATAC-seq-mode and ChIP-seq-mode peaks separately across 104 samples following the method of Meuleman et al. (2020), and joined the resulting peaks (when overlapping). This yielded a total of 976,813 reference peaks (excluding the Y Chromosome and unanchored scaffolds) with core and consensus segments (empirical confidence bounds of aggregates of peak summits and regions, respectively) (Supplemental File S1; Meuleman et al. 2020). Core and consensus segments amounted, respectively, to 134 Mb and 264 Mb, or 5.1% and 10.0% of genome space. Of these, 41,841 peaks (4.3%) were located in promotor regions (defined as 1 kb upstream of to 0.1 kb downstream from the transcription start sites [TSSs]) of 33,579 Ensembl reference transcripts (out of 43,512) and were referred to as “proximal,” whereas the remaining 934,972 peaks were considered “distal.” The median consensus size of the proximal peaks (306 bp) was larger than that of the distal peaks (216 bp; PWilcoxon < 2.2 × 10–16) (Fig. 1B). The proximal peaks were “open” in more tissues than the distal peaks (i.e., distal peaks were more often tissue-specific; PWilcoxon < 2.2 × 10–16) (Fig. 1C). The accessibility of “open” peaks was higher for proximal (14.0-fold increase of read depth over background) than for distal peaks (7.3-fold increase of read depth over background; PWilcoxon = 4.3 × 10–19) (Fig. 1D). Of note, the distribution of genomic evolutionary rate profiling (GERP) scores (Cooper et al. 2005; Davydov et al. 2010) was overdispersed for both proximal and distal peaks, showing an excess of positions with higher and lower substitution rates than expected (under neutrality) compared with flanking regions (Fig. 1E).
ATAC-seq peaks were unevenly distributed across the genome, both between and within chromosomes (Supplemental Fig. S4A,B). The density of ATAC-seq peaks was highest for Chromosome 19 and lowest for Chromosomes 6 and 12. Chromosome X was also particularly poor in ATAC-seq peaks, but this could be owing to its hemizygosity in a majority of male samples. The density of ATAC-seq peaks was highly correlated with the density of TSSs (r = 0.52) (Supplemental Fig. S4C).
We used unsupervised nonnegative matrix factorization (NMF) according to the method of Meuleman et al. (2020) to decompose the 976,813-peak × 104-sample matrix in 16 components (Fig. 1F; Supplemental Tables S5, S6; Supplemental File S1; Supplemental Figs. S5, S6). NMF converts each sample and each peak into a linear combination of these 16 components, that is, a weighted sum of the 16 components. Twelve of the 16 components could be readily assigned to recognizable bodily systems as they would be dominant (>30% of the weight) in anatomically related samples. They were labeled accordingly: central nervous system (CNS), cerebellum, immune system, digestive tract, ruminal epithelium, lower respiratory, upper respiratory, muscle, liver, endocrine, mammary gland, and testis. Accordingly, these 12 components dominated about 629,870 ATAC-seq peaks (64.5%) characterized by tissue-specific accessibility. Three components corresponded, respectively, to eight-cell embryo, morula, and inner cell mass (ICM) and dominated a very distinct set of about 213,305 ATAC-seq peaks (21.8%), of which 54,498 (5.6%), 76,175 (7.8%), and 14,108 (1.4%) were eight-cell, morula, and ICM specific, respectively. A set of 26,414 (2.7%) peaks was shared by multiple, yet at first glance, anatomically unrelated samples. The meaning of this 16th component, whether biological or technical, remains unclear. It is referred to as “undefined.” Of note, the 16th component dominated the sample types that were hard to dissociate. Finally, one group corresponded to about 107,224 (11.0%) peaks that were shared by the majority of samples and characterized by uniform weights for the 16 components (≤30% for any component). They are referred to as “ubiquitous” peaks and account for 59.6% of proximal peaks assigned to housekeeping genes (Supplemental Table S6).
NMF decomposition uses a binary matrix summarizing the presence (1) versus absence (0) of the 976,813 peaks in the 104 samples. We also performed hierarchical clustering of the samples (Ward D2 method) (R Core Team 2023) based on a quantitative measure of the accessibility of distal peaks. This approach grouped the samples largely by the NMF component (Supplemental Fig. S7). Of note, the cerebellar samples (assigned to the NMF07_Cerebellum group) formed a distinct cluster yet were closest to the remaining CNS samples (NMF01_CNS group). Also, the ruminal epithelial primary cells (NMF02_Ruminal epithelium) formed a distinct cluster, with embryonic samples (rather than digestive tract samples) as sister clade. This suggests that culturing these cells profoundly affects the epigenetic profile of these cells, apparently toward a proliferative stem-cell like phenotype.
We evaluated the added value of analyzing extra samples, first in terms of discovery of new ATAC-seq peaks. To that end, we ranked samples by the decreasing number of newly uncovered ATAC-seq peaks (Supplemental Fig. S8A). When limiting ourselves to the 97 postnatal tissue samples, the number of newly discovered peaks saturated at about 725,000 after approximately 75 samples, suggesting that our library of ATAC-seq peaks includes the majority of regulatory elements accessible after birth. However, adding only three embryonic samples (and, to a lesser extent, primary cultured cells) uncovered an extra tier of around 200,000 peaks. This suggests that substantially more developmental stage–specific regulatory elements remain to be uncovered and that the analysis of additional fetal samples, for instance, is warranted. An additional value in analyzing more tissue types is to determine in which tissue types known regulatory elements are accessible and in which tissues they are not (Supplemental Fig. S6). The majority of peaks uncovered in a given sample are neither unique for the sample nor shared with all others but rather shared with a variable number of other samples (not necessarily from the same NMF component) which are, hence, not obvious to predict (Supplemental Fig. S8B,C). Finally, we examined the relative merits of analyzing more sample types with ATAC-seq only versus fewer sample types with multiple assays. To that end, we evaluated the overlap between the peaks identified by Kern et al. (2021) in eight tissue types (adipose, cerebellum, brain cortex, hypothalamus, liver, lung, skeletal muscle, spleen) using ATAC-seq combined with ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF) with our own catalog. To make for a better comparison, we reanalyzed Kern's ATAC-seq data in “narrow-peak” mode (as opposed to the “broad-peak” mode used by Kern et al.). The vast majority (93.5%) of Kern's ATAC-seq peaks overlapped with ours, as expected. Kern's ATAC-seq peaks overlapped with 69% of their H3K4me3 peaks (i.e., active promotors), 43% of their H3K4me1 peaks (i.e., active enhancers), 44% of their H3K27ac peaks (i.e., active promotors and enhancers), and 42% of their CTCF peaks. Similarly, a subset of our ATAC-seq data from the corresponding eight tissues overlapped with 76% of H3K4me3 peaks, 50% of H3K4me1 peaks, 49% of H3K27ac peaks, and 48% of CTCF peaks. In comparison, our complete ATAC-seq peak catalog overlapped with 89% of H3K4me3 peaks, 73% of H3K4me1 peaks, 71% of H3K27ac peaks, and 69% of CTCF peaks (Supplemental Fig. S8D). Thus, it appears that analyzing more sample types by ATAC-seq compensates to some extent for the use of a single assay as it recovers regulatory elements that are missed if performing only ATAC-seq on fewer sample types. This finding also suggests that the same regulatory element may adopt distinct epigenetic configurations, presumably associated with distinct functional states, captured by distinct assays in different tissues.
We searched for TF binding motifs enriched in tissue-specific and ubiquitous ATAC-seq peaks using HOMER (Fig. 2A; Supplemental Tables S7, S8; Heinz et al. 2010). For each component, binding motifs were found to be very significantly enriched, in good agreement with previous reports for bovine tissue-specific cis-acting regulatory elements and/or tissue-specific function of the corresponding TF in other species. Moreover, using publicly available RNA-seq information, we found that 35 of the cognate TFs were more highly expressed in the corresponding tissues compared with all other ones (Supplemental Table S7).
Open chromatin regions are enriched in cis regulatory elements. (A) TFs (x-axis) whose binding motifs are enriched in tissue type–specific ATAC-seq peaks assigned to the corresponding NMF components (y-axis). The color code measures the excess in the percentage of peaks encompassing the corresponding motif over background, scaled (Z-score) across NMF components. TFs that are also more strongly expressed in tissues corresponding to that component compared with other tissues (Supplemental Table S7) are marked by asterisks. (B) Proportion of significant, across tissue type, correlations (P < 10–6) between ATAC-seq peak accessibility and gene expression as a function of the distance between the TSS and the peak. Green indicates positive correlations; red, negative correlations.
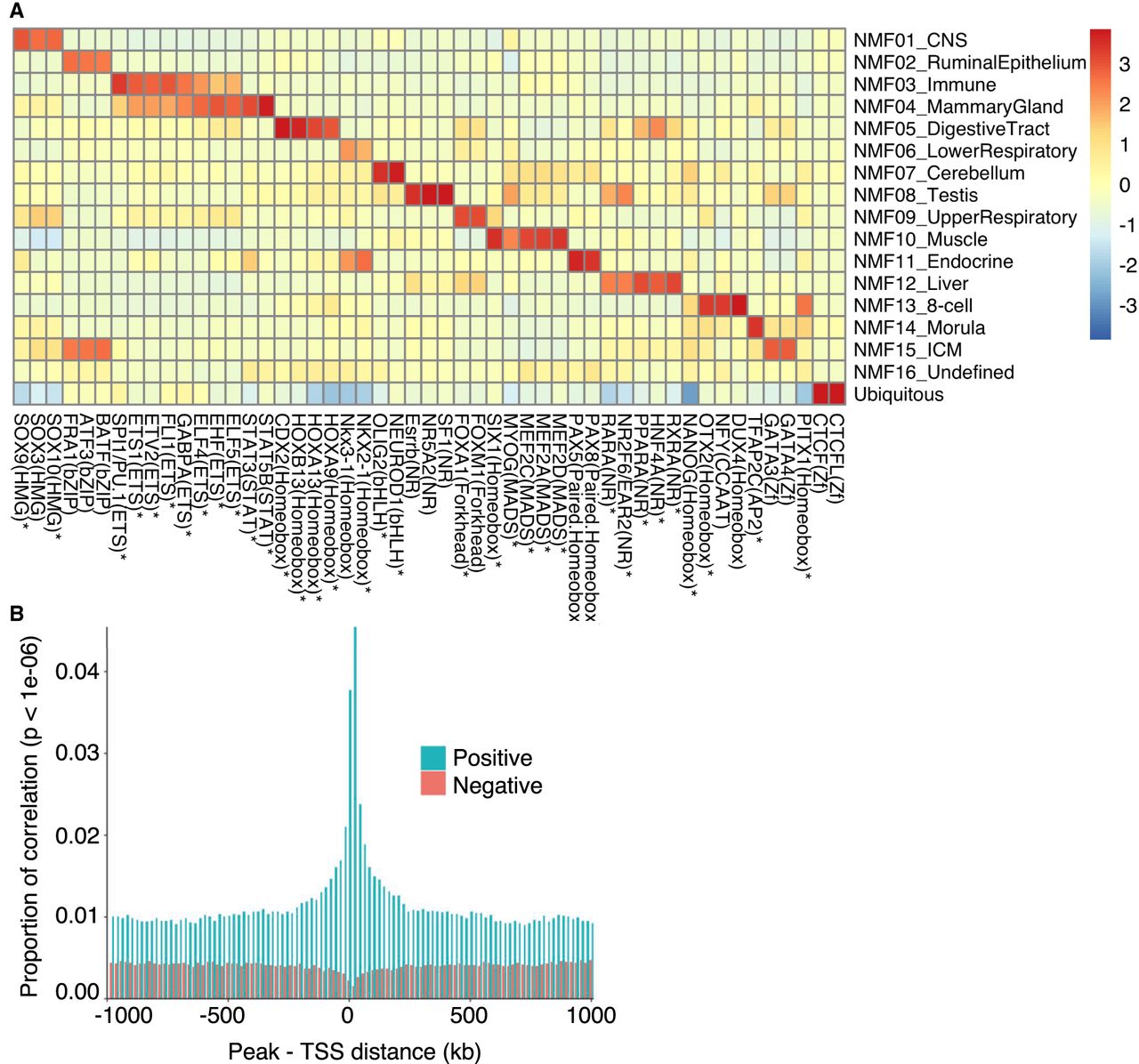
We matched 91 of our tissue-specific ATAC-seq data with publicly available RNA-seq data from 56 bovine tissues (Supplemental Table S9), and computed correlations between gene expression and accessibility of ATAC-seq peaks mapping within 1 Mb from the gene's TSS (Fig. 2B). Correlations were overdispersed, showing too many positive but also negative correlations. Indeed, any peak that is specific for a given tissue type will be positively correlated with any gene that is specifically expressed in that same tissue type. These correlations are therefore not indicative of cis interactions between peaks and their target gene(s). However, positive correlations were increasing in numbers (and becoming more significant) as the distance between peak and gene decreased. This inflation of positive correlations over the background (i.e., at distances ≥ 750 kb) was highly significant for gene-peak distances up to ∼250 kb. This supports the common occurrence of direct cis interactions between enhancer peaks and target genes, at least up to such distances. The effect was slightly more pronounced for peaks located downstream from the TSSs than peaks located upstream of the TSSs. The same trend was not observed when repeating the same analyses with negative correlations (Fig. 2B). In fact, we observed a slight deflation of negative correlations (becoming less negative and less significant) as the distance between the peak and gene decreased below ∼40 kb. This suggests that few ATAC-seq peaks act as cis silencers on target genes.
Generating a catalog of common variants mapping to cis-acting regulatory elements
We used a previously established catalog of 11,030,905 single-nucleotide variants (SNVs) and 1,705,738 short (≤265-bp) insertion–deletion variants (indels) obtained by analyzing 264 Holstein-Friesian (HF) whole-genome sequences (average, 25.2-fold depth; range, 15.2 to 47.1) (Oget-Ebrad et al. 2022) using GATK (Poplin et al. 2018). Of these, 1,256,997 SNVs (11.4%) and 133,394 indels (7.8%) mapped to ATAC-seq peaks (Supplemental File S2).
We studied the proportion of indels falling within versus outside ATAC-seq peaks separately for the following genome compartments: TSSs, 100 bp upstream of to 1 kb downstream from transcription termination sites (TTSs), exons, introns, and intergenic regions (Fig. 3B). The proportion of indels mapping to ATAC-seq peaks was significantly below expectations for TSSs (i.e., proximal peaks) (P = 1.4 × 10–33), TTSs (P = 1.2 × 10–52), introns (P < 1.0 × 10–100), and intergenic regions (P < 1.0 × 10–100). These effects were even stronger when considering common indels only (minor allele frequency [MAF] > 0.05). This is the expected signature of purifying selection acting on functionally important elements. Of note, the proportion of all indels (but not common indels) was slightly higher than expected (p = 0.18) for the exonic compartment. This effect became significant (P = 1.8 × 10–10) when restricting the analysis to open reading frames (ORFs) (i.e., ignoring 5′ and 3′ untranslated regions).
Open chromatin regions are mutational hotspots yet are subject to purifying selection. (A) Proportion of SNVs that map in ATAC-seq peaks for different genome compartments (x-axis: TSS, TTS, exon, intron, intergenic). Gray indicates the proportion of a corresponding genome compartment that is occupied by ATAC-seq peaks; dark purple, all SNVs; and light purple, common SNVs (MAF > 0.05). (***) P ≤ 0.001, (*) P ≤ 0.05. (B) As in A for indels. Gray indicates the proportion of a corresponding genome compartment that is occupied by ATAC-seq peaks; dark green, all indels; and light green, common indels (MAF > 0.05). (***) P ≤ 0.001, (NS) nonsignificant. (C) Number of singleton SNVs (per interrogated base pair) in 264 whole-genome-sequenced Holstein-Friesian (HF) animals in nonoverlapping 200-bp windows at increasing distances from the center of ATAC-seq peaks. The shaded area corresponds to 2 × SD for the corresponding window. Fluctuation increases with distance as the number of windows decreases. (D) As in C for singleton indels. The excess near the ATAC-seq peak centers is clearly visible despite the drop at their very center (assumed to reflect purifying selection). (E) Folded SFS (0.0 < MAF ≤ 0.1) for SNVs mapping within ATAC-seq peaks assigned to different genome compartments (purple range indicates TSS, TTS, exon, intron, intergenic) compared with SNVs outside peaks. (***) P ≤ 0.001. (F) Folded SFS (0.0 < MAF ≤ 0.1) for indels mapping within ATAC-seq peaks assigned to different genome compartments (green range indicates TSS, TTS, exon, intron, intergenic) compared with indels outside peaks. (***) P ≤ 0.001.
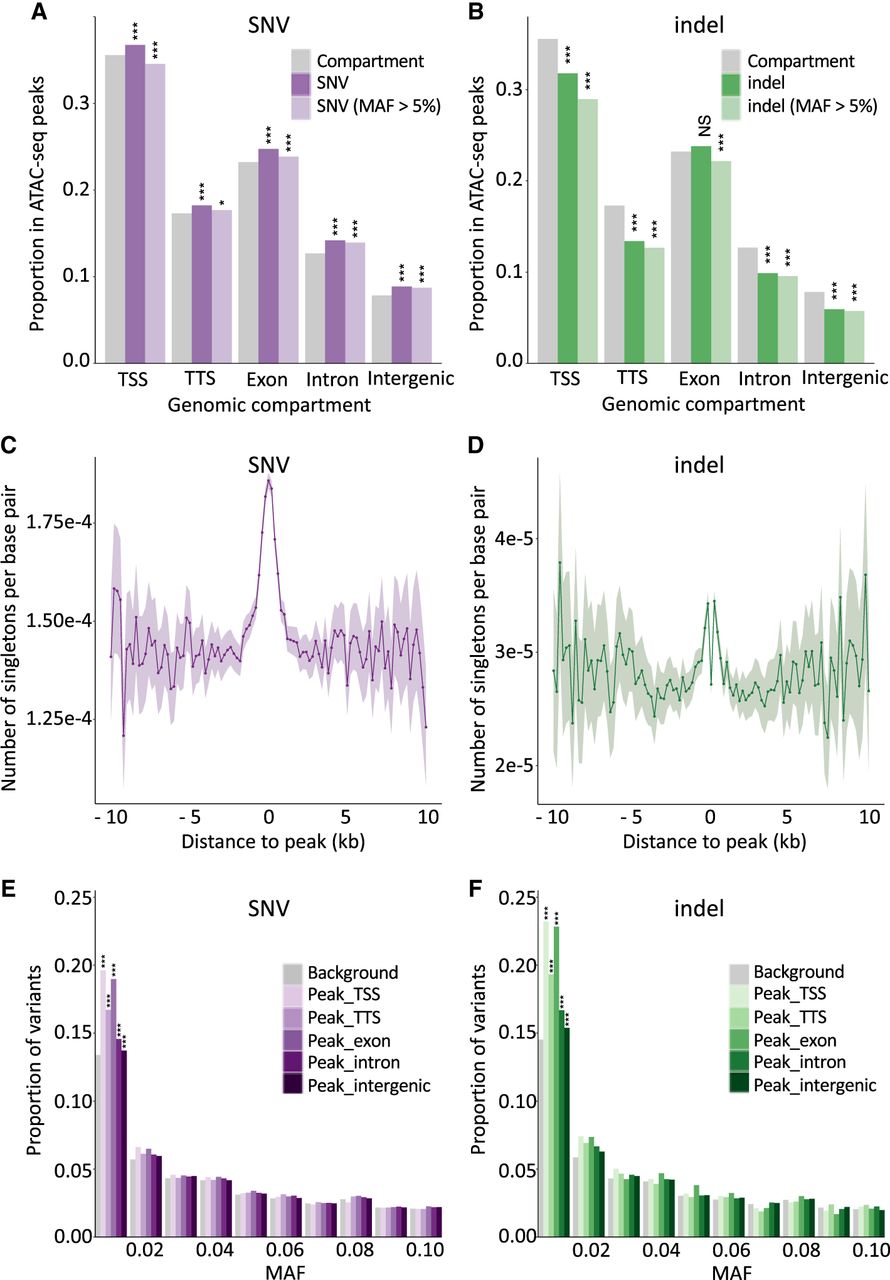
In contrast, the proportion of SNVs mapping to ATAC-seq peaks was significantly higher than expected for all five genomic compartments: TSSs (P = 2.7 × 10–21), TTSs (P = 3.9 × 10–19), exons (P = 4.4 × 10–41), introns (P < 1.0 × 10–100), and intergenic regions (P < 1.0 × 10–100) (Fig. 3A). The effect was reduced when considering common SNVs only but was still significant for TTSs (P = 2.6 × 10–3), exons (P = 8.7 × 10–6), introns (P < 10–100), and intergenic regions (P < 10–100). This is counter-intuitive as ATAC-seq peaks are assumed to be functionally important elements and, hence, subject to purifying selection that should result in fewer than expected number of variants. Only for TSSs were common SNVs significantly underrepresented in ATAC-seq peaks (P = 8.2 × 10–11). These observations corroborate recent findings in Arabidopsis thaliana (Monroe et al. 2022) and humans (Kaiser et al. 2021; Luquette et al. 2022). They may be related to the reduced efficiency of RNase H2–dependent repair of erroneously incorporated nucleotides during pol α-dependent initiation of DNA replication of Okazaki fragments (Reijns et al. 2015), or of nucleotide excision repair (Sabarinathan et al. 2016), at sites where proteins, including TFs, bind DNA. In agreement with this hypothesis, the density of singletons (supposed to be enriched in recent mutations and hence used as surrogate for de novo mutations [DNMs]) was higher in ATAC-seq peaks than in flanking sequences (Fig. 3C,D). Knowing that the expected number of singletons per base pair equals 4Nμ independently of sample size (Nielsen and Slatkin 2013), and under some simplifying assumptions, the SNV mutation rate may be about 1.3 times higher within than outside ATAC-seq peaks.
In melanoma, the rate of somatic mutations is increased about fivefold at accessible TF binding sites, and this is thought to be because of hampered nucleotide excision repair by bound TFs (Sabarinathan et al. 2016). To verify whether the excess of SNVs in our ATAC-seq peaks was likewise concentrated in TF binding motifs, we identified 386,812 NMF component-specific peaks (weight of one component >90%) and, within those, the positions of the 10 most enriched binding motifs for that component (de novo enrichment analysis) (Supplemental Table S8). We then checked whether SNVs mapping to the corresponding peaks would fall more often within than outside of the binding motifs. There was no evidence for a preferential location of SNVs in binding motifs, whether at the motif, NMF component, or global level. If any trend, the proportion of SNVs in binding motifs was slightly inferior to their corresponding peak occupancy (global P = 0.06) (Supplemental Table S10).
To further check whether variants mapping to ATAC-seq peaks, including SNVs, might be under purifying selection as expected, we compared the folded site frequency spectrum (SFS) of variants mapping within ATAC-seq peaks for the five genomic compartments in the 264 sequenced animals, with the folded SFS of all variants flanking peaks. The proportion with MAF ≤ 0.01 was higher for variants mapping in ATAC-seq peaks, and this applied both to indels and to SNVs. The effect was strongest for TSSs and exons (Fig. 3E,F).
Taken together, our data support the notion that ATAC-seq peaks are mutational hotspots, explaining the observed excess of SNVs, yet are subject to enhanced purifying selection, accounting for the depletion in indels and the shift of the SFS toward low frequencies for both SNVs and indels. This hypothesis may also account for the overdispersed GERP scores (Fig. 1E).
Of the 1,390,391 genetic variants mapping to ATAC-seq peaks, 847,831 SNVs and 86,673 indels are common with MAF > 0.05 in the sequenced animals (Supplemental File S2). These are prime candidates to receive particular attention when computing genomic breeding values in genomic selection.
Identifying bovine cis eQTL in liver and blood
To evaluate whether our catalog of “ATAC-seq variants” is enriched in regulatory variants, we performed eQTL analyses. We collected whole-blood and liver samples from, respectively, 224 and 176 HF cows and performed RNA-seq using standard procedures (Supplemental Table S11; Lee et al. 2021; Wathes 2021a,b). The reads were mapped to the bovine genome (ARS-UCD1.2) using HISAT2 (Kim et al. 2015) and read coverage for 27,233 reference genes (bosTau9.ensGene.gtf, v101) estimated using StringTie (Pertea et al. 2015). Gene count data were normalized within sample using DESeq2 (Love et al. 2014) following the method of Anders and Huber (2010) and across samples using inverse normal transformation. After filtering out lowly expressed genes, 14,289 genes were retained for eQTL analyses in blood and 15,458 in liver. All samples were genotyped with a high-density SNV array interrogating 777,962 variants and imputed to whole genome using Minimac4 (Das et al. 2016) and the 264 sequenced Holstein animals as reference. This yielded usable genotypes for 8.4 million SNVs and 1.3 million indels with MAF > 0.02. Cis eQTL analyses (variants within 1 Mb from gene's TSS) were conducted using residuals corrected for hidden PEER factors (Stegle et al. 2010), country (of origin of the samples), and polygenic effects estimated with GenABEL (Aulchenko et al. 2007), under an additive model using QTLtools (Delaneau et al. 2017). Nominal P-values were corrected for multiple testing within the 2-Mb cis window by permutation. The best-corrected P-value was retained for each gene and converted to FDR value by tissue type. Cis eQTLs with FDR < 0.05 were considered significant.
We obtained 7817 significant cis eQTLs in blood and 6172 in liver (Supplemental Table S12). These numbers correspond to 39.9% and 54.7% of interrogated genes, respectively, and are comparable to findings in humans (https://gtexportal.org/home/tissueSummaryPage). Leading variants tended to concentrate (and −log(p) values hence to be highest) in the vicinity of the TSSs (Supplemental Fig. S9). The proportion of significant blood eQTLs that would also operate in liver was estimated at 67% using π1 following the method of Storey and Tibshirani (2003), whereas the proportion of significant liver eQTLs that would also operate in blood was estimated at 78%.
We defined “credible variant sets” (i.e., sets of variants that are more likely to include the causative variants that are functionally driving the observed cis eQTL effect) as the leading variant plus the variants in LD with it at threshold r2-value of 0.9. The median size of credible sets was 12, ranging from one to 2870.
Variants driving eQTL are preferentially mapping in ATAC-seq peaks
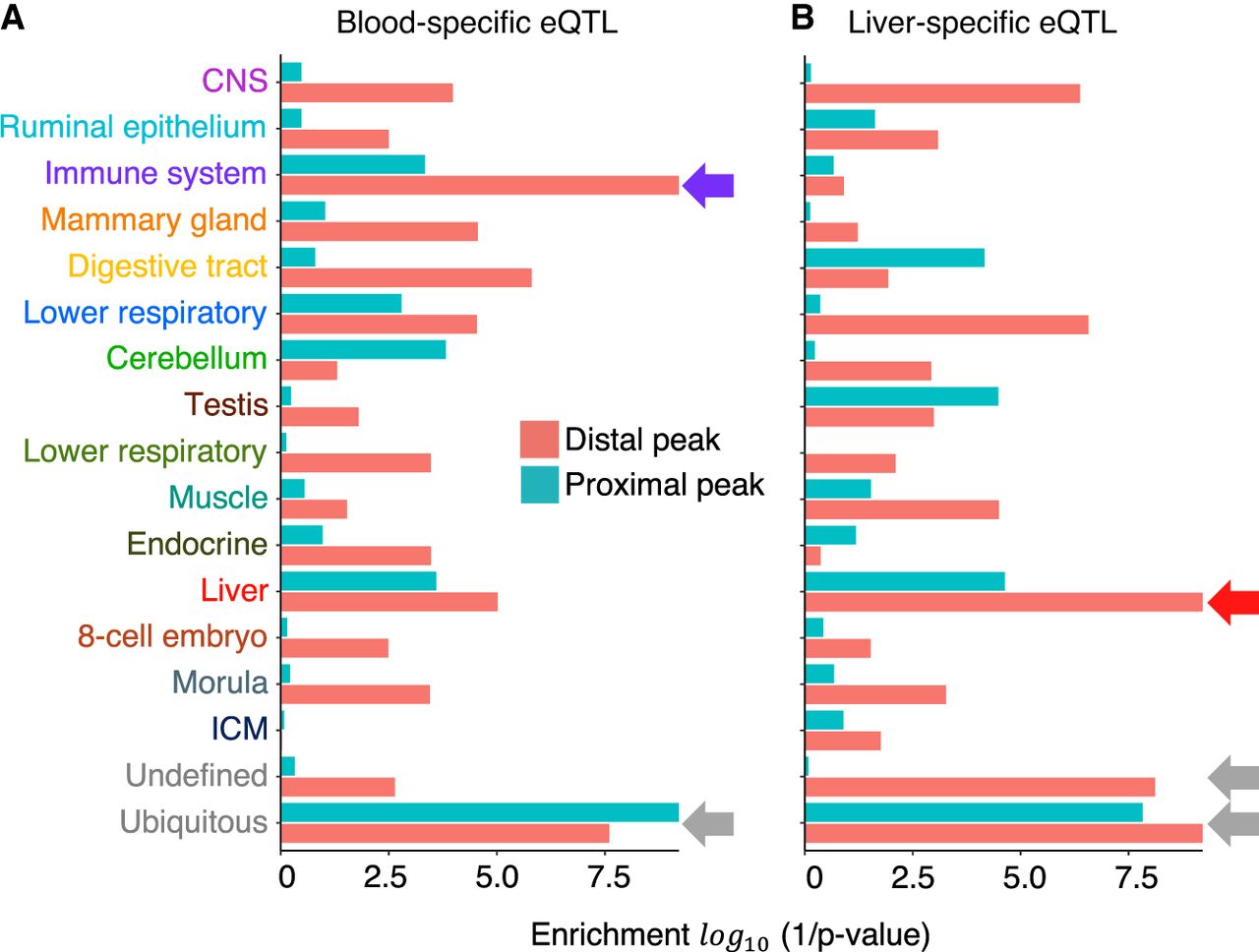
If variants mapping to ATAC-seq peaks are indeed enriched in causative variants, they should be enriched in the credible sets driving cis eQTL effects. The significance of the overlap between cis eQTL credible sets and ATAC-seq peaks was evaluated by permutation following the method of Trynka et al. (2015) (Fig. 4A,B; Supplemental Table S13). Analyses were conducted by NMF component (assigning peaks to their dominant component). Credible sets for blood-specific cis eQTLs were most significantly (P ≤ 0.0001) enriched in variants mapping to ATAC-seq peaks assigned to the immune and ubiquitous NMF components. Credible sets for liver-specific cis eQTLs were most significantly (P ≤ 0.0001) enriched in variants mapping to ATAC-seq peaks assigned to the liver and ubiquitous NMF components. The enrichment in tissue-specific ATAC-seq peaks (immune for blood eQTL, and liver for liver eQTL) was driven by distal peaks, whereas the enrichment in ubiquitous ATAC-seq peaks was driven by both proximal and distal peaks (Fig. 4A,B; Supplemental Table S13).
Open chromatin regions are enriched in cis-regulatory variants. Enrichment of variants mapping to NMF component-specific ATAC-seq peaks in credible sets (r2 ≥ 0.9 with the lead variant) of 3857 blood-specific and 2212 liver-specific cis eQTL, evaluated by following the method of Trynka et al. (2015). The x-axis shows statistical significance (log(1/p)) of the enrichment; y-axis, NMF component. Green indicates proximal peaks; red, distal peaks. (A) Blood-specific eQTLs. (B) Liver-specific eQTLs.
Estimating the proportion of regulatory variants mapping in ATAC-seq peaks and the proportion of variants mapping in ATAC-seq peaks that are regulatory
The utility of ATAC-seq data for the identification of regulatory variants underpinning the heritability of complex traits depends on the proportion of regulatory variants that map to ATAC-seq peaks (i.e., the sensitivity or ratio of true positives/[true positives + false negatives]), and the proportion of regulatory variants among variants mapping to ATAC-seq peaks (i.e., the precision or ratio of true positives/[true positives + false positives]). The combination of ATAC-seq and cis eQTL information provides an opportunity to estimate these parameters. For example, if all cis eQTLs are driven by a regulatory variant mapping to an ATAC-seq peak, all credible sets should contain at least one variant mapping to an ATAC-seq peak. We developed a maximum likelihood-based approach (see Methods) to estimate the proportion of cis eQTLs driven by regulatory variants in ATAC-seq peaks from the observed excess of credible set variants mapping in ATAC-seq peaks (over the proportion of the genome occupied by ATAC-seq peaks). The parameters estimated by this approach were then used to estimate the proportion of regulatory variants among those that map to ATAC-seq peaks (see Methods).
We applied this approach to the 7817 blood and 6172 liver eQTLs. It yielded estimates of 0.34 (blood) and 0.32 (liver) for sensitivity, and 0.044 (blood) and 0.041 (liver) for precision. In other words, approximately one out of three regulatory variants maps to an ATAC-seq peak, and approximately one in 25 variants mapping to ATAC-seq peaks is regulatory.
Retrospective evaluation of the utility of ATAC-seq information for the identification of known regulatory variants in livestock
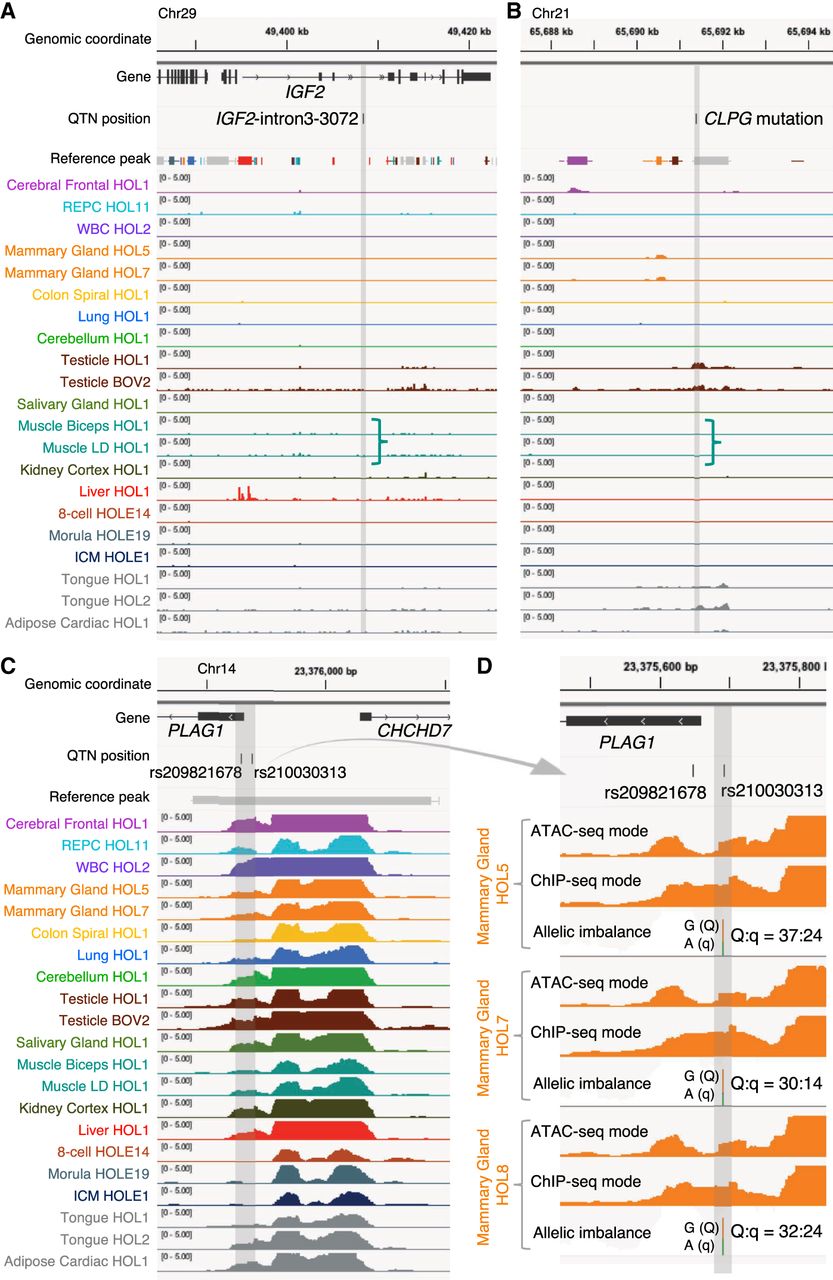
Prior positional cloning studies conducted in livestock identified at least three regulatory variants influencing economically important quantitative traits. The first is the IGF2-intron3-3072 variant in the pig that precludes binding of the ZBED6 repressor to a conserved silencer element in intron 3 of the IGF2 gene, leading to illegitimate postnatal expression of the paternal IGF2 allele in striated muscle and, hence, muscular hypertrophy (Van Laere et al. 2003; Markljung et al. 2009). The sequence conservation of the corresponding silencer element suggests that it operates in a similar manner across species. Nevertheless, there was no evidence in our ATAC-seq peak catalog of any peak overlapping the orthologous position of the quantitative trait nucleotide (QTN; bosTau9 Chr 29: 49,408,409), whether tissue specific (including in muscle) or tissue shared (Fig. 5A). The second is the ovine rs10721113 callipyge QTN that perturbs the function of a putative silencer element highly conserved among placental mammals, located in the GTL2-DLK1 intergenic region, that—in wild-type sheep—suppresses postnatal muscular expression of a cluster of imprinted genes (including the paternally expressed DLK1 and PEG11 genes). Animals inheriting this mutation from their sire express the callipyge muscular hypertrophy (Freking et al. 2002; Georges et al. 2004). There was no clear evidence of a peak overlapping the orthologous position of the QTN (bosTau9 Chr 21: 65,691,395) in postnatal skeletal muscle ATAC-seq peaks, as would be expected. There was such a peak in testes and to a lesser extend in tongue, but—in hindsight—this would not have been considered strong support for the causality of the corresponding variant, and the significance of this finding—if any—remains unclear (Fig. 5B). The third example concerns a credible set of eight noncoding variants affecting bovine stature and several other traits by perturbing the expression of PLAG1 and possibly other genes in its vicinity (Karim et al. 2011). Of the eight variants, only the two that map to the supposedly bidirectional promotor between PLAG1 and CHCHD7 (rs209821678: (CCG)9/(CCG)11 microsatellite and rs210030313: A/G SNV) overlap with strong ubiquitous ATAC-seq peaks (Fig. 5C). Of note, previously conducted reporter and EMSA assays supported the causality of both variants (Karim et al. 2011). Further supporting their causality, the ATAC-seq data reveal an allelic imbalance for the rs210030313 SNV (Fig. 5D) that is consistent with the observed effects on gene expression (G = Q allele = higher PLAG1/CHCHD7 expression = more accessible; A = q allele = lower PLAG1/CHCHD7 expression = less accessible). Moreover, the rs209821678 variant lies in a trough revealed in the ATAC-seq mode profile, suggesting that the corresponding segments mediated binding to a trans-acting factor. In this case, ATAC-seq data would therefore have been helpful in pinpointing the causative variants.
A retrospective evaluation of the utility of the ATAC-seq catalog for identifying regulatory variants. ATAC-seq peaks at three genomic loci encompassing regulatory QTNs previously identified in domestic animals. Chromosome coordinates, gene annotations, QTN positions, core and consensus reference peak regions (thick bars and horizontal lines, respectively; color-coded based on their highest NFM component), and peaks (ChIP-seq mode tag coverage unless otherwise mentioned) from at least one tissue sample representing each NMF component group with corresponding color code. Positions of the QTNs are highlighted as vertical gray bands. Track height measures the normalized tag coverage (1,000,000/[total tag count]). (A) The bovine orthologous region encompassing the IGF2-intron3-3072 QTN identified in pigs (A/G at susScr11: Chr 2: 1,483,817; bosTau9: Chr 29: 49,408,408) (Van Laere et al. 2003; Markljung et al. 2009) that maps to a 16-bp motif highly conserved among placental mammals disrupts interaction of the ZBED6 repressor, resulting in an approximately threefold up-regulation of IGF2 in postnatal skeletal muscle affecting muscle growth, heart size, and fat deposition. None of ATAC-seq peaks overlapping the 16-bp motif were called across the 104 ATAC-seq data analyzed in this study. (B) The bovine orthologous region encompassing the callipyge (CLPG) muscular hypertrophy mutation identified in sheep (A/G at oviAri4: Chr 18: 64,294,536; bosTau9: Chr 21: 65,691,397) (Freking et al. 2002; Smit et al. 2003). The mutation is located in a 12-bp highly conserved motif among placental mammals and is considered to disrupt a muscle-specific long-range control element (a silencer) that causes ectopic expression of a 327-kb cluster of imprinted genes in postnatal skeletal muscle. ATAC-seq peaks overlapping the mutation site were called only in testis and tongue samples but not in skeletal muscle. (C) Bovine PLAG1 promoter region encompassing two out of eight candidate QTNs influencing bovine stature identified by Karim et al. (2011) (rs209821678 [alternatively ss319607405]: (CCG)11/(CCG)9 at bosTau9: Chr 14: 23375648–23375650; rs210030313 [ss319607406]: G/A at bosTau9: Chr 14: 23375692). The two QTNs reside in a strong 1044-bp-long ubiquitous peak between the PLAG1 and CHCHD7 TSSs. Regions encompassing the other six credible variants do not map to any called peak in our ATAC-seq data and, hence, are not shown. (D) Enlargement of the two QTN loci for three animals that are Qq heterozygous at rs210030313. Peaks called with ATAC-seq and ChIP-seq modes, as well as allelic imbalance in mapped reads, are shown. The two QTNs reside in a footprint of the ATAC-seq mode peak, which is recovered by a ChIP-seq mode peak, indicating the presence of trans-acting factor(s) in the region hindering cleavage events by transposases. Allelic imbalance at rs210030313 (Q = G; q = A) indicates that the Q allele is more accessible compared with the q allele. Previous work showed that the two regulatory variants affect bidirectional promoter strength and that the Q allele, associated with bigger stature, showed approximately 1.5-fold higher promoter activity compared with the q allele in a luciferase assay. Figures were created using the Integrative Genomics Viewer (Robinson et al. 2011).
Discussion
We herein report the most complete catalog of open chromatin regions for cattle to date (Fig. 1; Supplemental File S1; e.g., Foissac et al. 2019; Halstead et al. 2020a,b; Kern et al. 2021; Ming et al. 2021). It comprises more than 976,000 ATAC-seq peaks detected in one or more of 63 tissue types representing pregastrulation embryos, endoderm, ectoderm, and mesoderm. To facilitate its use by the community, the data are made accessible via a custom track on the UCSC Genome Browser (via https://genome.ucsc.edu/s/Animal_Genomics_ULiege/ATAC_hub_V1 or https://www.gigauag.uliege.be/cms/c_4791343/en/gigauag-diagnostics-software-data). The vast majority of ATAC-seq peaks (about 840,000) show tissue-specific accessibility, dominated by one of 16 NMF components (weight of the largest NMF component >0.3). Of note, nearly 213,000 of these are specific for preimplantation embryonic stages. This clearly indicates that, as expected, chromatin accessibility is very dynamic, warranting the analyses of multiple tissues during fetal development in future studies. By studying across-tissue correlation between gene expression (using publicly available RNA-seq data) and accessibility of neighboring peaks, we show a clear signal of enhancer and/or promotor activity (excess of positive correlations with decreasing distance) but not of silencer activity (depletion of negative correlations with decreasing distance). This either indicates that silencers only account for a small minority of cis-acting regulatory elements or that silencers are not effectively identified using assays relying on chromatin openness (see also hereafter).
One of the main motivations to establish open chromatin catalogs in livestock is to identify regulatory variants that might underpin the heritability of agronomically important traits. Indeed, it is hoped that knowledge of these regulatory variants may increase the accuracy of genomic selection. We identified 1,390,391 variants mapping to open chromatin regions, of which 938,374 (67%) are common variants with MAF > 0.05 in Dutch HFs (Supplemental File S2). Instead of prioritizing variants mapping to ATAC-seq peaks indiscriminately for genomic selection, our catalog can be used to define sets of variants that are accessible in tissue types that are relevant for the trait under consideration. For instance, variants mapping to ATAC-seq peaks that are specifically accessible in, for instance, the mammary gland, hypothalamus, pituitary gland, and liver might be particularly relevant when targeting milk production traits.
We note that the proportion of SNVs (as opposed to indels) mapping to ATAC-seq peaks is significantly higher than the proportion of genome space occupied by ATAC-seq peaks (Fig. 3). We provide evidence that this is owing to an approximately 1.3-fold higher DNM rate in ATAC-seq peaks compared with the rest of the genome, corroborating recent findings in other eukaryotes (Kaiser et al. 2021; Luquette et al. 2022; Monroe et al. 2022). Shifts toward lower MAFs compared with variants in flanking regions support the operation of purifying selection on open chromatin regions and, hence, their functional importance (Fig. 3).
To further examine the regulatory function of open chromatin regions, we identified 7817 and 6172 sets of credible variants assumed to include causative variants driving the same number of cis eQTLs detected in blood and liver, respectively (Supplemental Table S12). As anticipated, variants in these credible sets tend to map to open chromatin regions more often than expected by chance alone (as evaluated by permutation following the method of Trynka et al. 2015). Furthermore, the enrichment was not random with respect to the NMF component (Fig 4; Supplemental Table S13). Credible sets corresponding to blood eQTLs tended to overlap ATAC-seq peaks assigned to the immune and ubiquitous NMF, whereas liver eQTLs tended to overlap ATAC-seq peaks assigned to the liver and ubiquitous NMF. The overlap with the tissue-specific NMF (immune and liver) was primarily owing to distant regulatory elements, whereas the overlap with the ubiquitous NMF was equally owing to distant and proximal regulatory elements (Fig. 4).
These results tell us that variants that map to ATAC-seq peaks are more likely to be regulatory variants than variants that map outside of ATAC-seq peaks. However, they do not really tell us how sensitive and precise ATAC-seq assays are to identify regulatory variants. We summarized this interrogation with two specific questions: (1) what fraction of regulatory variants map to ATAC-seq peaks (sensitivity), and (2) what fraction of variants in ATAC-seq peaks are regulatory (precision). We developed a maximum likelihood framework using eQTL information to estimate both parameters. Sensitivity was estimated at one in three, and precision at one in 25. Thus, as many as two out of three regulatory variants may lie outside of ATAC-seq peaks inventoried in our catalog (Supplemental File S2). A first possible explanation of this observation is that our catalog still misses a substantial proportion of bovine gene switches. This could be because, in particular, we did not explore sufficient developmental stages, or ATAC-seq peaks do not capture all gene switches (e.g., silencers). A second possible explanation is that variants lying outside of ATAC-seq peaks may nevertheless affect the functionality of (nearby) gene switch components that are identified by ATAC-seq peaks (e.g., by affecting the formation of secondary structures involving the switch). Finally, some variants are known to affect transcript levels not by perturbing gene switches but by affecting transcript stability, including stop gains and splice variants. For example, the K232A mutation in DGAT1 affects transcript abundance by affecting splicing (Fink et al. 2020). The one in 25 precision indicates that the majority of variants falling in ATAC-seq peaks are probably neutral. It is possible that some eQTLs are driven by more than one causative variant, which would slightly increase precision. Contrary to coding variants, which can be identified quite accurately based on our understanding of the genetic code and splicing mechanisms, predicting the effect of SNVs on the functionality of cis-acting regulatory elements is still in its infancy.
Taken together, our results indicate that the knowledge of open chromatin regions in the bovine genome is a first step toward the identification of regulatory variants, yet this knowledge will likely have to be complemented with additional information to more effectively pinpoint the causative regulatory variants and thereby have a major impact on the accuracy of genomic selection.
Methods
Ethical approvals, sample collection, and processing
All relevant procedures using animals were approved by the animal care and use committee (ACUC) of the University of Liège (approval no. 17-1948 and 17-1949) or the Ruakura ethics committee, Hamilton, New Zealand (approval no. AEC 12845) and performed in accordance with the relevant guidelines and regulations of the committees. Blood samples were collected from the tail or jugular vein of animals using K2-EDTA blood collection tubes. White blood cells (WBCs) were enriched by lysing erythrocytes using eBioscience 10X RBC lysis buffer (Thermo Fisher Scientific). Peripheral blood mononuclear cells (PBMCs) were prepared by density gradient centrifugation using either Ficoll-Paque plus (Cytiva) or Lymphoprep (Stemcell Technologies) with SepMate-50 tubes (Stemcell Technologies). For tissue collections, animals were humanely euthanized, and tissues were collected in ice-cold Belzer UW cold storage solution (Bridge to Life) until processing as described below or were otherwise snap-frozen in liquid nitrogen. Details of bovine samples can be found in Supplemental Tables S1 and S2. To optimize protocols, one C57BL/6J × A/J F1 mouse (male, 1 yr old) was euthanized by cervical dislocation. Four tissues (liver, spleen, kidney, muscle) were collected in the UW cold storage solution and processed as described below. We processed/stored samples in three ways: fresh, slow-frozen, and snap-frozen conditions.
Fresh samples
Tissues collected in the UW cold storage solution were directly used for constructing ATAC-seq libraries on the day of sampling (17 biosamples with “fresh” in their names) (Supplemental Table S3).
Cryopreserved samples
Tissues were cut into ∼27-mm3 cubes and transferred to cryotubes filled with 1 mL of STEM-CELLBANKER DMSO-free cell freezing media (Amsbio). The tubes were kept on ice for ∼10 min and transferred to a cryo-box in dry ice while other samples were processed. The samples were then stored in a −80°C freezer until use (55 biosamples with “slow” in names) (Supplemental Table S3).
Snap-frozen samples
Tissues were cut into ∼27-mm3 cubes and transferred to cryotubes. The samples were snap-frozen in liquid nitrogen and stored at −80°C until use (34 biosamples with “snap” in names) (Supplemental Table S3).
ATAC-seq library construction
ATAC-seq libraries were constructed following the Omni ATAC-seq protocol (Corces et al. 2017) with some modifications.
Tissue homogenization
A cryopreserved tissue in a vial was quickly thawed in a water bath and transferred to an excess amount of ice-cold Dulbecco's phosphate buffered saline (DPBS). The cryopreserved or fresh tissue samples were dissociated into a single-cell suspension using a gentleMACS dissociator (Miltenyi Biotec) by running one or two cycles of program B1 with 3 mL of ice-cold Omni 1 × homogenization buffer in a gentleMACS C-tube. Snap-frozen samples were pulverized using a mortar and pestle chilled in liquid nitrogen. The cell suspension or pulverized tissue was transferred to a Dounce tissue grinder (Merck D9063) on ice with 3 mL of ice-cold Omni 1 × homogenization buffer. Samples were homogenized with an A-pestle until resistance went away and further with a B-pestle (three to 10 strokes each) so as to disrupt cellular plasma membranes. Cell debris were removed by passing the sample through stackable cell strainers (100-, 70-, and 30-µm MACS SmartStrainers, Miltenyi Biotec). The flow-through was further clarified by a brief centrifugation at 100g for 1 min at 4°C. The supernatant was mixed with an equal volume of ice-cold Omni 50% iodixanol solution (final, 25% iodixanol).
Purification of nuclei
Two layers of iodixanol cushions were prepared in a 2-mL LoBind tube (Eppendorf) by placing 600 µL of ice-cold 40% iodixanol solution on the bottom (marking the surface of the bottom layer facilitated sample collection later) and overlaying 600 µL of ice-cold 29% iodixanol solution using a wide-bore tip. On the top, 800 µL of the cell suspension (containing 25% iodixanol) was placed. Density gradient centrifugation was performed using a table-top centrifuge (Eppendorf 5430R) with a swing rotor at 6000g for 30 min at 4°C and a soft brake setting. Top layers were carefully removed down to ∼2 mm above the bottom layer. The nuclear fraction, between the bottom and middle layers, was collected (∼400 µL) to a new LoBind tube on ice. The number of nuclei was counted by mixing 20 µL of the sample with 20 µL of 100 × diluted Hoechst 33342 (Thermo Fisher Scientific) using a hemocytometer under a fluorescence microscope.
Tagmentation
Approximately 50,000 nuclei were transferred to two 1.5-mL LoBind tubes filled with 1 mL of ice-cold Omni-ATAC-RSB + 0.1% Tween-20 buffer and centrifuged at 500g for 10 min at 4°C. After carefully removing the supernatant, nuclei were resuspended in 50 µL of Omni-ATAC reaction mix containing Tn5 transposase TDE1 enzyme (Illumina). As the effectiveness of transposase varied slightly among samples, we used two different amounts of the enzyme per sample (Supplemental Table S3). After mixing the sample by pipetting six times using a P200 fine tip, tagmentation reaction was performed using an Eppendorf ThermoMixer at 500 rpm for 30 min at 37°C. The reaction was stopped by adding 300 µL of PB buffer in a MinElute PCR purification kit (Qiagen) and 10 µL of 3 M sodium acetate (pH 5.2). The sample was mixed, kept at room temperature for 10 min, and stored at −20°C until DNA purification. Libraries for two blood samples (WBC, PBMC) were generated with an alternative ATAC-seq protocol (Buenrostro et al. 2013) during the pilot experiment phase. A genomic DNA (gDNA) control library was also prepared using 50 ng of purified gDNA from one animal (HOL1_m) by following the Nextera DNA sample preparation guide (Illumina). The tagmented DNA was purified and eluted in 21 µL elution buffer using the MinElute PCR purification kit.
Library preparation
The purified DNA was amplified using NEBNext high-fidelity 2X PCR master mix with the Ad1 and Ad2 primers (Buenrostro et al. 2013) for 13 (for ATAC-seq library) or five PCR-cycles (gDNA library), respectively. The amplified libraries were purified and eluted in 50 µL elution buffer using the MinElute PCR purification kit. Library size distribution was monitored using 10 µL of the library by QIAxcel capillary electrophoresis (Qiagen). Large DNA fragments were eliminated using AMPure XP magnetic beads (Beckman Coulter) by a right-side size selection using 0.55 × followed by 1.5 × volume ratio of beads to sample. Library concentration was estimated using the KAPA library quantification kit (Kapa Biosystems). The libraries were sequenced with 2 × 38-bp paired-end reads using a NextSeq 500 sequencer, or 2 × 51-bp paired-end reads on a NovaSeq 6000 (Illumina) instrument. In total, 185 ATAC-seq libraries were sequenced, yielding 31.8 million paired-end fragments on average (range: 2.5–117.2 million fragments) (Supplemental Table S3). ATAC-seq FASTQ files were obtained from the EMBL-EBI ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) under accession number E-MTAB-9872 (Lee et al. 2021) or generated in this study and submitted under accession numbers E-MTAB-11825 and E-MTAB-11826 (see Data access). In addition, we downloaded publicly available ATAC-seq data from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA531214, PRJNA665194, PRJNA601200, PRJNA595394, and PRJNA622966 (Supplemental Table S1; Fang et al. 2019; Halstead et al. 2020a,b; Johnston et al. 2021) and analyzed these in a similar way.
ATAC-seq peak calling
ATAC-seq peaks were called following the recommendations of the ENCODE ATAC-seq pipeline (“ATAC-seq Data Standards and Processing Pipeline”) (https://www.encodeproject.org/atac-seq/).
Trimming
Sequences with low sequence quality, residuals of library adaptors, and bases >38 bp (to uniform read length across data) were trimmed using Trimmomatic (ILLUMINACLIP:NexteraPE-PE.fa:2:30:5:1:true SLIDINGWINDOW:4:15 MINLEN:20 CROP:38) (Bolger et al. 2014). Proportions of reads remaining after trimming averaged 98.7% (range: 96.8–99.5%) (Supplemental Table S3).
Mapping
The trimmed reads were aligned to the bovine genome assembly ARS-UCD1.2 using Bowtie 2 (‐‐local –mm). Overall mapping rate averaged 95.5% (range: 36.4%–99.4%).
Filtration
Reads mapping to the mitochondrial chromosome were filtered out using SAMtools (samtools idxstats file.bam | cut -f 1 | grep -v chrM | xargs samtools view -b file.bam) (Danecek et al. 2021). The proportion of mitochondrial reads averaged 16.1% (range: 0.8%–61.9%). PCR/optical duplicates were removed using Picard toolkit (java -jar picard.jar MarkDuplicates REMOVE_DUPLICATES = true OPTICAL_DUPLICATE_PIXEL_DISTANCE = 100 [2500 for NovaSeq data] VALIDATION_STRINGENCY = LENIENT) (http://broadinstitute.github.io/picard/). Duplicate read rate averaged 12.7% (range: 4.9%–23.1%). Properly aligned reads with high sequencing quality were selected with SAMtools (for paired-end reads, samtools view -f 3 -F 1284 -q 30; for single-end reads after trimming, samtools view -f 9 -F 260 -q 30), resulting in an average of 35.3 million informative reads per library (range: 3.4–141.5 million).
Fractionation
Reads were partitioned into two bins based on their mapped fragment lengths using BamTools filter function (Barnett et al. 2011): (1) short reads generated from putative nucleosome-free regions of DNA (<146 bp) and (2) longer reads likely from nucleosome-associated DNA (≥146 bp).
Peak calling
ATAC-seq peaks were called using MACS2 in two ways (Supplemental Fig. S3): First, in ATAC-seq mode: the genomic locus cleaved by the transposase (a tag) was defined as a 38-bp region centered either 4 bp (for a plus strand read) or 5 bp (for a minus strand read) downstream from the read's 5′-end (Adey et al. 2010; Buenrostro et al. 2013). Peaks (open chromatin regions) were identified by comparing the tag distribution of a sample to one from a purified gDNA control (macs2 callpeak ‐‐format BED ‐‐control ‐‐nomodel ‐‐shift -19 ‐‐extsize 38 ‐‐qvalue 0.05 ‐‐gsize hs ‐‐keep-dup all ‐‐max-gap 38 ‐‐SPMR ‐‐bdg). Second, in ChIP-seq mode, to recover regions protected from transposase cleavage events owing to binding of trans-regulatory factor(s) like a TF (so-called footprints in the ATAC-seq mode analysis), peaks were called using general settings used for ChIP-seq analysis (MACS2 piles up entire sequencing fragments instead of focusing on transposase cleavage sites close to 5′-ends of reads). To avoid covering nucleosome positions, only the nucleosome-free fraction of sequence fragments (mapped fragment size < 146 bp) was used (macs2 callpeak ‐‐format BAMPE ‐‐control ‐‐qvalue 0.05 ‐‐gsize hs ‐‐keep-dup all ‐‐max-gap 38) for ChIP-seq mode.
Quality control
In-house ATAC-seq data (peak-called with the ATAC-seq mode) were evaluated using commonly used ATAC-seq quality-control measurements (Supplemental Table S3): the fraction of reads in called peak regions (FRiP; average: 0.212; range: 0.004–0.591) and TSS enrichment (average: 16.1; range: 2.14–46.2). Irreproducible discovery rates (IDRs) that measure reproducibility in score ranking between peaks, as well as rescue ratios that measure consistency between replicates (average: 1.23; range: 1.02–1.96), were calculated using samples with technical replicates.
Low-quality libraries with the number of filtered reads of fewer than 10 million per sample, FRiP less than 0.07, TSS enrichment less than 7.0, and a self-consistency ratio more than two, as well as some technical duplicates with less quality, were excluded from further analyses (36 libraries out of 185).
Final peak calling per biosample
Reads of libraries from the same biosample that passed the quality control were merged, and peaks were called afresh as described above in ATAC-seq and ChIP-seq modes. We also integrated 15 high-quality public data sets in our analysis (Fang et al. 2019; Halstead et al. 2020a,b; Johnston et al. 2021). P-value thresholds for final peak selection for all samples (with and without technical replicates) were determined as the median of the lowest log(1/P)-values of peaks with IDR ≤ 0.1 across samples with technical replicates (−log10(P-value) = 8.01 and 9.28 for ATAC-seq and ChIP-seq modes, respectively).
Reproducibility
Reproducibility of peak calling was evaluated by measuring Pearson's correlation of genome-wide read coverage in 500-bp windows between technical (range: 0.89–0.99) and biological replicates (0.85–0.97) (Supplemental Figs. S1, S2) using deepTools’ bamCoverage (‐‐outFileFormat bigwig ‐‐effectiveGenomeSize 2701495761 ‐‐normalizeUsing RPKM ‐‐ignoreForNormalization chrX chrY), multiBigwigSummary (bins ‐‐binSize 500), and plotCorrelation (‐‐corMethod pearson ‐‐whatToPlot heatmap –removeOutliers ‐‐colorMap viridis ‐‐plotNumbers) (Ramírez et al. 2016).
Defining and merging consensus and core peak components across samples and calling modes
Core and consensus peak components were defined following the method of Meuleman et al. (2020) as follows. Individual peak (IP) summit positions were collated across samples separately for ATAC-seq and ChIP-seq modes. Summits were clustered such that the distance between clusters was ≥20 bp. The space covered by each cluster was defined as the core component of a newly defined “collective peak” (CP). The corresponding IP were piled up and the limits of the consensus CP defined as the full-width at half maximum. If by doing so some consensus CPs overlapped, they were merged by repeating the process using all concerned IPs. This yielded two genome-wide sets of core and consensus CPs for ATAC-seq mode and ChIP-seq mode, respectively. If overlapping, the corresponding consensus and core CPs were merged to, in the end, yield one unique set of core and consensus “CPs” or reference peaks used for all further analyses (Supplemental File S1).
Nonnegative matrix factorization
NMF was conducted following the method of Meuleman et al. (2020) using scripts downloaded from GitHub (https://github.com/Altius/Vocabulary). Briefly, we set up an m (number of samples) × n (number of peaks) matrix (V) summarizing the accessibility of each peak in each sample in binary mode (0 or 1, based on presence absence of an “IP” in the corresponding sample). V was decomposed in a m × k W and k × n H matrix such that V ≈ W × H, where k is the number of hidden components. The value for k was set at 16 following the method of Meuleman et al. (2020), as a trade-off between maximizing the recapitulation of V and retaining biological interpretability (k at elbow point of the derivative of F1 score over k). Following this procedure, each sample and each peak were assigned a weight for each one of the k components. Samples and peaks were in general assigned to their dominant component (with largest score), and components were assigned to biological systems on the basis of their composite samples (Fig. 1F; Supplemental Table S5; Supplemental File S1). The peak information is also made accessible via a custom track on the UCSC Genome Browser (https://genome.ucsc.edu/s/Animal_Genomics_ULiege/ATAC_hub_V1 or https://www.gigauag.uliege.be/cms/c_4791343/en/gigauag-diagnostics-softwar e-data).
Public RNA-seq data
RNA-seq data originated from bovine tissues similar to the ones generated in this study were downloaded from the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra; for accession numbers, see Supplemental Table S9; Graf et al. 2014; Cai et al. 2018; Dado-Senn et al. 2018; Khansefid et al. 2018; Fang et al. 2019, 2020). Low-quality bases, residuals of library adaptors, and short reads <35 bp were removed using Trimmomatic (ILLUMINACLIP:adaptor.fa:2:30:10:4:true SLIDINGWINDOW:4:15 LEADING:10 TRAILING:10 MINLEN:36). The reads were mapped to the bovine reference genome ARS-UCD1.2 using HISAT2 (version 2.1.0) using genome indexes that were built along with coordinates of 2.7 million DNA variants (2,389,896 SNVs and 176,799 indels) and reference transcripts (bosTau9.ncbiRefSeq.gtf; hisat2 ‐‐dta ‐‐no-softclip -x index -S out.sam). Reads mapped to ribosomal RNA, duplicated, or improperly mapped were filtered out using BEDTools (intersectBed -abam -b rRNA.bed -v), Picard toolkit (MarkDuplicates REMOVE_DUPLICATES = true VALIDATION_STRINGENCY = LENIENT), and SAMtools (view -f 3 -F 1284 -q 30). Expression levels of reference transcripts on autosomes and sex chromosomes in the gene reference file (bosTau9.ensGene.gtf, v101) were estimated using StringTie (stringtie -G -e). A raw read count matrix per gene (27,233 genes) was prepared using a prepDE.py script in StringTie. Using R package DESeq2, the count data were normalized by their library sizes after selecting genes for which counts were more than 10 (for the TF binding motif enrichment analysis) or 30 (for the peak-expression correlation analysis) in at least one sample (Data < −Data [(rowSums (counts (Data)) > threshold) ≥ 1]) and transformed using regularized-logarithm transformation (rlog (Data, blind = TRUE)).
TF binding motif enrichment in peaks
Known and de novo DNA motifs enriched in core peaks assigned to tissue specific components (one weight ≥ 0.9) or the ubiquitous component (all weights ≤ 30%) were identified using HOMER (findMotifsGenome.pl peak.bed bosTau9_genome_directory-size given) (Fig. 2A; Supplemental Tables S7, S8). For each of the 16 components, we then checked, for the 10 most enriched binding motifs, whether the cognate TF was also more strongly expressed in the tissue samples assigned to that component. This was accomplished by standardizing (Z-score) the expression level of the corresponding TF gene across 114 of the above-mentioned publicly available RNA-seq libraries that could be assigned to one of our 16 components (Supplemental Table S7), and verifying whether the Z-scores were higher in the tissue type assigned to the cognate component compared with the other samples. The statistical significance of the difference in Z-score was estimated using a permutation test and ensuing P-values converted to FDR values (π0 set at 1) using the qvalue R package (http://github.com/jdstorey/qvalue) to correct for multiple testing. FDRs ≤ 0.05 were deemed significant.
Correlation between chromatin openness and gene expression
Chromatin openness
Chromatin openness of a peak in a given sample was measured as the fold enrichment (in normalized read depth) over gDNA background at the nucleotide position in the peak with the highest such value. This was computed with the MACS2 bdgcmp function (-m FE -t sample_pileup.bdg -c control_lambda.bdg) and using the bedGraph files from MACS2 ATAC-seq mode peak calling. The highest fold enrichment value per peak was extracted using BEDTools (map -nonamecheck -c 4 -o max -a consensus_peak.bed -b out.bdg). We kept peaks for which fold enrichments were more than five at least in one sample (975,488 peaks).
Gene expression
Fifty-six bovine public RNA-seq data matched to 91 of our 104 ATAC-seq data sets were selected from the data sets mentioned above (Supplemental Table S9). RNA-seq data were processed as described above.
Correlation
Pearson's correlations between openness (fold enrichment) of peaks located within 1 Mb from TSS of a given gene and gene expression level across the 91 data sets were calculated using R stats (R Core Team 2023).
Generating a catalog of common variants mapping to cis-acting regulatory elements
Genome-wide variant catalog
We used a catalog of 11,030,905 SNVs and 1,705,738 short (≤265 bp) indels called from whole-genome sequences of 264 HF cattle (obtained from BioProject accession number PRJEB53518; Oget-Ebrad et al. 2022).
Proportion of variants mapping to ATAC-seq peaks by genome-compartment
The genome was subdivided in five mutually exclusive compartments (TSS, TTS, exonic, intronic, intergenic) using a gene reference file (bosTau9.ensGene.gtf, v101). Each compartment was further subdivided in (1) a part overlapping any peak in our catalog of 948,566 autosomal consensus peaks and (2) the remaining part. We then checked whether there was a significant difference in the proportion of variants mapping to the peak part versus the proportion of space occupied by the peak part using a chi-square goodness-of-fit test.
Density of singletons within and outside of ATAC-seq peaks
The change of singleton density as a function of the distance from the nearest peak was determined sequentially as follows. We first identified the size of the genome (in base pairs) that was within 100 bp from the nearest ATAC-seq peak (200-bp windows centered on the peaks; g1), as well as the number of singletons that mapped within this space (s1), and computed the corresponding ratio (r1 = s1/g1). We then identified the size of the genome that was between 300 and 100 bp from the nearest ATAC-seq peaks (200-bp windows on the left and right of window 1, excluding what was assigned to fraction 1; g2L and g2R), as well as the number of singletons that mapped within these spaces (s2L and s2R), and computed the corresponding ratios (r2L = s2L/g2L and r2R = s2R/g2R). We pursued this process for windows that were more and more distant from the nearest ATAC-seq peaks. The “confidence interval” around the estimates was defined as the computed ratio ±2 SD (2 × SDi), where SDi was computed assuming a binomial distribution as .
Site frequency spectrum
Variants mapping to peaks were sorted according to the five above-mentioned compartments (TSS, TTS, exonic, intronic, intergenic). Their SFS (0.01 bins) was compared (histogram) between compartments and with that of all other variants in the genome. To check for a shift toward lower allelic frequencies by compartment, we compared the distribution of allelic frequencies between variants that mapped to peaks (“peak part” above) versus variants that did not map to peaks (but belonged to the same compartment) using a Wilcoxon rank-sum test.
Identifying bovine cis eQTLs in liver and blood
RNA-seq and data preprocessing
We reanalyzed RNA-seq data of 176 liver and 227 whole-blood biopsies collected from 240 Holstein females at ∼14 d postpartum in the GplusE project (obtained from ArrayExpress; accession numbers E-MTAB-9347 and E-MTAB-9431 for blood; E-MTAB-9348 and E-MTAB-9871 for liver) (Lee et al. 2021; Wathes et al. 2021a,b). The libraries were constructed with an Illumina TruSeq stranded total RNA library prep Ribo-Zero gold kit and sequenced with 75-base single-end reads. First, low-quality bases, residuals of library adaptors, and short reads (<35 bp) were removed using Trimmomatic (java -jar trimmomatic-0.36.jar SE input.fastq.gz output_trimmed.fastq.gz ILLUMINACLIP:TruSeq3-SE.fa:2:30:10 SLIDINGWINDOW:4:15 LEADING:3 TRAILING:3 MINLEN:36 2>>log.txt). The reads were mapped to the bovine reference genome ARS-UCD1.2 using HISAT2 (hisat2 ‐‐dta ‐‐no-softclip -x index -U trimmed.fastq.gz -S output.sam ‐‐rna-strandness R 2>>log.txt). Reads mapped on ribosomal RNA were filtered out using SAMtools (samtools sort input.sam -o sorted.bam) and BEDTools (intersectBed -abam sorted.bam -b rRNA.bed -v). BAM files from the same biosample were merged, and properly mapped reads were kept with SAMtools (samtools merge merged.bam sample_ID*.bam; samtools view -F 2308 -q 30 -o clean.bam -b merged.bam; samtools sort -o clean.sorted.bam clean.bam; samtools index clean.sorted.bam). Expression levels of reference transcripts (bosTau9.ensGene.gtf, v101) on autosomes and sex chromosomes were estimated using StringTie (stringtie clean.sorted.bam ‐‐rf -G bosTau9.ensGene.nochrUn.gtf -e -o transcripts.gtf) (Pertea et al. 2015). A raw read counts matrix by gene (27,233 genes) was prepared using a prepDE.py script in StringTie. Gene-specific reads counts were scaled with a “size factor” using DESeq2 after eliminating mitochondrial gene counts. Gene with summation of TPM lower than one and with fewer than eight individuals with counts greater than zero were filtered out. Afterward, counts were normalized by inverse normal transformation by gene and across individuals.
SNV genotyping
All animals were genotyped with a high-density (about 778,000) SNV array (Illumina BovineHD Genotyping BeadChip), and imputed to whole genome using SHAPEIT4 (for phasing) (Delaneau et al. 2019) and Minimac4 using the previously mentioned whole-genome sequences of 264 HF animals as reference. Variants with MAF ≤ 0.02, probability of the data assuming Hardy–Weinberg equilibrium ≤0.001, and imputation accuracy (r2) ≤ 0.9 were filtered out, leaving a total of 10,257,878 usable markers (the genotypes are available from the Zenodo open data repository at https://doi.org/10.5281/zenodo.8339268).
eQTL analyses
We used QTLtools to ensure RNA–DNA sample matching based on genotype concordance (Supplemental Table S11). Expression values were first corrected for hidden confounders and “country of origin” using probabilistic estimation of expression residuals (PEER). The resulting residuals were then further corrected for stratification and/or polygenic effects on gene expression using GenABEL. The ensuing “double-corrected” residuals were then used for cis eQTL analyses using QTLtools. For each gene, we tested all variants within 1 Mb from the TSS. Ensuing P-values were corrected for multiple testing (in the window) by permutation. For each gene, we kept the best P-value (= “lead variant”), and these “best P-values” were converted to FDR and Q-values (hence corrected for multiple testing) following the methods of Benjamini and Hochberg (1995) and Storey and Tibshirani (2003), respectively. eQTL with FDR ≤ 0.05 were deemed experiment-wide significant. π1 (the proportion of alternative hypotheses among all tested hypotheses) was estimated according to the method of Storey and Tibshirani (2003).
Enrichment of eQTL driving variants in ATAC-seq peaks
We first identified, for each significant cis eQTL, a credible set of variants defined as all the variants within 1 Mb from the lead variant and in LD with it with a threshold value . We then used the method proposed by Trynka et al. (2015) to measure the putative enrichment of credible variants in ATAC-seq peaks. The analysis was performed by NMF component. Briefly, we defined, for each cis eQTL, a region/window spanned by the credible set plus buffer segments on either side corresponding to twice the median peak size (= 436 bp). We first counted, using the real eQTL results, for how many eQTLs at least one credible variant mapped within an ATAC-seq peak (assigned to the NMF component under consideration). We then randomly shifted variant and peak coordinates with respect to each other within each cis eQTL window and counted for how many eQTLs at least one credible variant mapped within an ATAC-seq peak. This “permutation” process was repeated 10,000 times, and the significance of the overlap between credible variants and peaks observed for the real data was evaluated from the number of occurrences of an equal or higher overlap with the permuted data. To evaluate the contribution of proximal rather than distal ATAC-seq peaks to the signal, permutations were also conducted separately by peak type (proximal vs. distal).
Estimating the proportion of regulatory variants mapping in ATAC-seq peaks and the proportion of variants mapping in ATAC-seq peaks that are regulatory
We assumed that every cis eQTL i out of T is driven by one regulatory variant that is part of a credible set comprising niA variants in the ATAC-seq peaks and niN variants outside of the ATAC-seq peaks. We further assumed that a fraction fA of cis eQTLs is driven by a regulatory variant mapping to an ATAC-seq peak (Supplemental Codes S1, S2), as well as a fraction fN = 1 − fA by a regulatory variant mapping outside ATAC-seq peaks, and that ATAC-seq peaks occupy a proportion pA of the genome. The likelihood of the data for eQTL i can hence be expressed as
We used the Newton–Raphson method (R nlm function) (R Core Team 2023) to determine the value of fA that maximizes the likelihood of the data for all T eQTL:
Data access
ATAC-seq data generated in this study have been submitted to the EMBL-EBI ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) under accession numbers E-MTAB-11825 and E-MTAB-11826. Imputed genotypes of animals used for eQTL analyses are available from the Zenodo open data repository at https://doi.org/10.5281/zenodo.8339268. Other data sets used in this study, published previously, are described in the Methods. Key analysis pipelines are available at GitHub (https://github.com/can11sichuan/Bov-ATAC) and Supplemental Code S1 and S2. The UCSC Genome Browser track hub to visualize all 104 individual and reference ATAC-seq peaks is accessible from https://genome.ucsc.edu/s/Animal_Genomics_ULiege/ATAC_hub_V1 or https://www.gigauag.uliege.be/cms/c_4791343/en/gigauag-diagnostics-software-data. Bovine ATAC-seq peaks and putative regulatory variants identified in this study are found in Supplemental Files S1 and S2, respectively.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Calixte Bayrou, Ken Kusakabe, Ruth Appeltant, Anne-Sophie Van Laere, and all members of Michel Georges’ laboratory for their help for sample collections, technical support, and fruitful discussion. We also thank the support provided by the GIGA Genomics and Bioinformatics core facilities. This work was funded by the Damona European Research Council advanced grant from the EU (AdG-GA323030), the GplusE FP7 grant from the EU (no. 613689), the CAUSEL grant from the Walloon Region (no. 1710030), and financial support from Inoveo. C.C. and T.D. are senior research associate and research director from the Fonds de la Recherche Scientifique. Computational resources have been provided by the Consortium des Équipements de Calcul Intensif, funded by the Fonds de la Recherche Scientifique de Belgique (no. 2.5020.11) and by the Walloon Region.
Author contributions: H.T., M.G., T.D., C.C., D.C.W., and M.A.C. conceived and supervised the project. G.C., A.S., G.C.M.M., C.C., Z.C., M.S., and M.L. contributed to sample and data collections. H.T. and L.T. performed wet-laboratory experiments. V.A.P., C.O.-E., G.C.M.M., J.L.G., T.L., W.C., and T.D. assisted data analysis. C.Y., H.T., and M.G. performed data analysis. M.G., H.T., and C.Y. wrote the manuscript.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277947.123.
[3] School of Veterinary Medicine, University College Dublin, Dublin 4, Ireland
References
- ↵Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, 2010. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11: R119. 10.1186/gb-2010-11-12-r119
- ↵Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. 10.1186/gb-2010-11-10-r106
- ↵Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. 2007. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23: 1294–1296. 10.1093/bioinformatics/btm108
- ↵Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT. 2011. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27: 1691–1692. 10.1093/bioinformatics/btr174
- ↵Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x
- ↵Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170
- ↵Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688
- ↵Cai W, Li C, Liu S, Zhou C, Yin H, Song J, Zhang Q, Zhang S. 2018. Genome wide identification of novel long non-coding RNAs and their potential associations with milk proteins in Chinese Holstein cows. Front Genet 9: 281. 10.3389/fgene.2018.00281
- ↵Cooper GM, Stone EA, Asimenos G, NISC Comparative Sequencing Program, Green ED, Batzoglou S, Sidow A. 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15: 901–913. 10.1101/gr.3577405
- ↵Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, 2017. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14: 959–962. 10.1038/nmeth.4396
- ↵Dado-Senn B, Skibiel AL, Fabris TF, Zhang Y, Dahl GE, Peñagaricano F, Laporta J. 2018. RNA-seq reveals novel genes and pathways involved in bovine mammary involution during the dry period and under environmental heat stress. Sci Rep 8: 11096. 10.1038/s41598-018-29420-8
- ↵Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, 2021. Twelve years of SAMtools and BCFtools. Gigascience 10: giab008. 10.1093/gigascience/giab008
- ↵Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, 2016. Next-generation genotype imputation service and methods. Nat Genet 48: 1284–1287. 10.1038/ng.3656
- ↵Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6: e1001025. 10.1371/journal.pcbi.1001025
- ↵Delaneau O, Ongen H, Brown AA, Fort A, Panousis NI, Dermitzakis ET. 2017. A complete tool set for molecular QTL discovery and analysis. Nat Commun 8: 15452. 10.1038/ncomms15452
- ↵Delaneau O, Zagury J-F, Robinson MR, Marchini JL, Dermitzakis ET. 2019. Accurate, scalable and integrative haplotype estimation. Nat Commun 10: 5436. 10.1038/s41467-019-13225-y
- ↵The ENCODE Project Consortium, Abascal F, Acosta R, Addleman NJ, Adrian J, Afzal V, Ai R, Aken B, Akiyama JA, Jammal OA, 2020. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583: 699–710. 10.1038/s41586-020-2493-4
- ↵Fang L, Liu S, Liu M, Kang X, Lin S, Li B, Connor EE, Baldwin RL, Tenesa A, Ma L, 2019. Functional annotation of the cattle genome through systematic discovery and characterization of chromatin states and butyrate-induced variations. BMC Biol 17: 68. 10.1186/s12915-019-0687-8
- ↵Fang L, Cai W, Liu S, Canela-Xandri O, Gao Y, Jiang J, Rawlik K, Li B, Schroeder SG, Rosen BD, 2020. Comprehensive analyses of 723 transcriptomes enhance genetic and biological interpretations for complex traits in cattle. Genome Res 30: 790–801. 10.1101/gr.250704.119
- ↵Fink T, Lopdell TJ, Tiplady K, Handley R, Johnson TJJ, Spelman RJ, Davis SR, Snell RG, Littlejohn MD. 2020. A new mechanism for a familiar mutation: Bovine DGAT1 K232A modulates gene expression through multi-junction exon splice enhancement. BMC Genomics 21: 591. 10.1186/s12864-020-07004-z
- ↵Foissac S, Djebali S, Munyard K, Vialaneix N, Rau A, Muret K, Esquerré D, Zytnicki M, Derrien T, Bardou P, 2019. Multi-species annotation of transcriptome and chromatin structure in domesticated animals. BMC Biol 17: 108. 10.1186/s12915-019-0726-5
- ↵Freking BA, Murphy SK, Wylie AA, Rhodes SJ, Keele JW, Leymaster KA, Jirtle RL, Smith TPL. 2002. Identification of the single base change causing the callipyge muscle hypertrophy phenotype, the only known example of polar overdominance in mammals. Genome Res 12: 1496–1506. 10.1101/gr.571002
- ↵García-Ruiz A, Cole JB, VanRaden PM, Wiggans GR, Ruiz-López FJ, Van Tassell CP. 2016. Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc Natl Acad Sci 113: E3995–E4004. 10.1073/pnas.1519061113
- ↵Georges M, Charlier C, Smit M, Davis E, Shay T, Tordoir X, Takeda H, Caiment F, Cockett N. 2004. Toward molecular understanding of polar overdominance at the ovine callipyge locus. Cold Spring Harb Symp Quant Biol 69: 477–484. 10.1101/sqb.2004.69.477
- ↵Graf A, Krebs S, Zakhartchenko V, Schwalb B, Blum H, Wolf E. 2014. Fine mapping of genome activation in bovine embryos by RNA sequencing. Proc Natl Acad Sci 111: 4139–4144. 10.1073/pnas.1321569111
- ↵Halstead MM, Kern C, Saelao P, Wang Y, Chanthavixay G, Medrano JF, Van Eenennaam AL, Korf I, Tuggle CK, Ernst CW, 2020a. A comparative analysis of chromatin accessibility in cattle, pig, and mouse tissues. BMC Genomics 21: 698. 10.1186/s12864-020-07078-9
- ↵Halstead MM, Ma X, Zhou C, Schultz RM, Ross PJ. 2020b. Chromatin remodeling in bovine embryos indicates species-specific regulation of genome activation. Nat Commun 11: 4654. 10.1038/s41467-020-18508-3
- ↵Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004
- ↵Johnston D, Kim J, Taylor JF, Earley B, McCabe MS, Lemon K, Duffy C, McMenamy M, Cosby SL, Waters SM. 2021. ATAC-seq identifies regions of open chromatin in the bronchial lymph nodes of dairy calves experimentally challenged with bovine respiratory syncytial virus. BMC Genomics 22: 14. 10.1186/s12864-020-07268-5
- ↵Kaiser VB, Talmane L, Kumar Y, Semple F, MacLennan M, Deciphering Developmental Disorders Study, FitzPatrick DR, Taylor MS, Semple CA. 2021. Mutational bias in spermatogonia impacts the anatomy of regulatory sites in the human genome. Genome Res 31: 1994–2007. 10.1101/gr.275407.121
- ↵Karim L, Takeda H, Lin L, Druet T, Arias JAC, Baurain D, Cambisano N, Davis SR, Farnir F, Grisart B, 2011. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet 43: 405–413. 10.1038/ng.814
- ↵Kern C, Wang Y, Xu X, Pan Z, Halstead M, Chanthavixay G, Saelao P, Waters S, Xiang R, Chamberlain A, 2021. Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research. Nat Commun 12: 1821. 10.1038/s41467-021-22100-8
- ↵Khansefid M, Pryce JE, Bolormaa S, Chen Y, Millen CA, Chamberlain AJ, Vander Jagt CJ, Goddard ME. 2018. Comparing allele specific expression and local expression quantitative trait loci and the influence of gene expression on complex trait variation in cattle. BMC Genomics 19: 793. 10.1186/s12864-018-5181-0
- ↵Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317
- ↵Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923
- ↵Lee Y-L, Takeda H, Costa Monteiro Moreira G, Karim L, Mullaart E, Coppieters W, The GplusE consortium, Appeltant R, Veerkamp RF, Groenen MAM, 2021. A 12 kb multi-allelic copy number variation encompassing a GC gene enhancer is associated with mastitis resistance in dairy cattle. PLoS Genet 17: e1009331. 10.1371/journal.pgen.1009331
- ↵Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478: 476–482. 10.1038/nature10530
- ↵Liu X, Li YI, Pritchard JK. 2019. Trans effects on gene expression can drive omnigenic inheritance. Cell 177: 1022–1034.e6. 10.1016/j.cell.2019.04.014
- ↵Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8
- ↵Luquette LJ, Miller MB, Zhou Z, Bohrson CL, Zhao Y, Jin H, Gulhan D, Ganz J, Bizzotto S, Kirkham S, 2022. Single-cell genome sequencing of human neurons identifies somatic point mutation and indel enrichment in regulatory elements. Nat Genet 54: 1564–1571. 10.1038/s41588-022-01180-2
- ↵Markljung E, Jiang L, Jaffe JD, Mikkelsen TS, Wallerman O, Larhammar M, Zhang X, Wang L, Saenz-Vash V, Gnirke A, 2009. ZBED6, a novel transcription factor derived from a domesticated DNA transposon regulates IGF2 expression and muscle growth. PLoS Biol 7: e1000256. 10.1371/journal.pbio.1000256
- ↵Meuleman W, Muratov A, Rynes E, Halow J, Lee K, Bates D, Diegel M, Dunn D, Neri F, Teodosiadis A, 2020. Index and biological spectrum of human DNase I hypersensitive sites. Nature 584: 244–251. 10.1038/s41586-020-2559-3
- ↵Ming H, Sun J, Pasquariello R, Gatenby L, Herrick JR, Yuan Y, Pinto CR, Bondioli KR, Krisher RL, Jiang Z. 2021. The landscape of accessible chromatin in bovine oocytes and early embryos. Epigenetics 16: 300–312. 10.1080/15592294.2020.1795602
- ↵Monroe JG, Srikant T, Carbonell-Bejerano P, Becker C, Lensink M, Exposito-Alonso M, Klein M, Hildebrandt J, Neumann M, Kliebenstein D, 2022. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 602: 101–105. 10.1038/s41586-021-04269-6
- ↵Nielsen R, Slatkin M. 2013. An introduction to population genetics: theory and applications. Oxford University Press, Oxford, New York.
- ↵Oget-Ebrad C, Kadri NK, Moreira GCM, Karim L, Coppieters W, Georges M, Druet T. 2022. Benchmarking phasing software with a whole-genome sequenced cattle pedigree. BMC Genomics 23: 130. 10.1186/s12864-022-08354-6
- ↵Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33: 290–295. 10.1038/nbt.3122
- ↵Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, 2018. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 10.1101/201178
- ↵Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. 10.1093/nar/gkw257
- ↵R Core Team. 2023. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Reijns MAM, Kemp H, Ding J, de Procé SM, Jackson AP, Taylor MS. 2015. Lagging-strand replication shapes the mutational landscape of the genome. Nature 518: 502–506. 10.1038/nature14183
- ↵Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754
- ↵Sabarinathan R, Mularoni L, Deu-Pons J, Gonzales-Perez A, López-Bigas N. 2016. Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature 532: 264–267. 10.1038/nature17661
- ↵Smit M, Segers K, Carrascosa LG, Shay T, Baraldi F, Gyapay G, Snowder G, Georges M, Cockett N, Charlier C. 2003. Mosaicism of solid gold supports the causality of a noncoding A-to-G transition in the determinism of the callipyge phenotype. Genetics 163: 453–456. 10.1093/genetics/163.1.453
- ↵Stegle O, Parts L, Durbin R, Winn J. 2010. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol 6: e1000770. 10.1371/journal.pcbi.1000770
- ↵Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440–9445. 10.1073/pnas.1530509100
- ↵Trynka G, Westra H-J, Slowikowski K, Hu X, Xu H, Stranger BE, Klein RJ, Han B, Raychaudhuri S. 2015. Disentangling the effects of colocalizing genomic annotations to functionally prioritize non-coding variants within complex-trait loci. Am J Hum Genet 97: 139–152. 10.1016/j.ajhg.2015.05.016
- ↵Van Laere A-S, Nguyen M, Braunschweig M, Nezer C, Collette C, Moreau L, Archibald AL, Haley CS, Buys N, Tally M, 2003. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425: 832–836. 10.1038/nature02064
- ↵Wathes DC, Becker F, Buggiotti L, Crowe MA, Ferris C, Foldager L, Grelet C, Hostens M, Ingvartsen KL, Marchitelli C, 2021a. Associations between circulating IGF-1 concentrations, disease status and the leukocyte transcriptome in early lactation dairy cows. Ruminants 1: 147–177. 10.3390/ruminants1020012
- ↵Wathes DC, Cheng Z, Salavati M, Buggiotti L, Takeda H, Tang L, Becker F, Ingvartsen KI, Ferris C, Hostens M, 2021b. Relationships between metabolic profiles and gene expression in liver and leukocytes of dairy cows in early lactation. J Dairy Sci 104: 3596–3616. 10.3168/jds.2020-19165
- ↵Xiang R, van den Berg I, MacLeod IM, Hayes BJ, Prowse-Wilkins CP, Wang M, Bolormaa S, Liu Z, Rochfort SJ, Reich CM, 2019. Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. Proc Natl Acad Sci 116: 19398–19408. 10.1073/pnas.1904159116
- ↵Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, 2008. Model-based Analysis of ChIP-seq (MACS). Genome Biol 9: R137. 10.1186/gb-2008-9-9-r137