
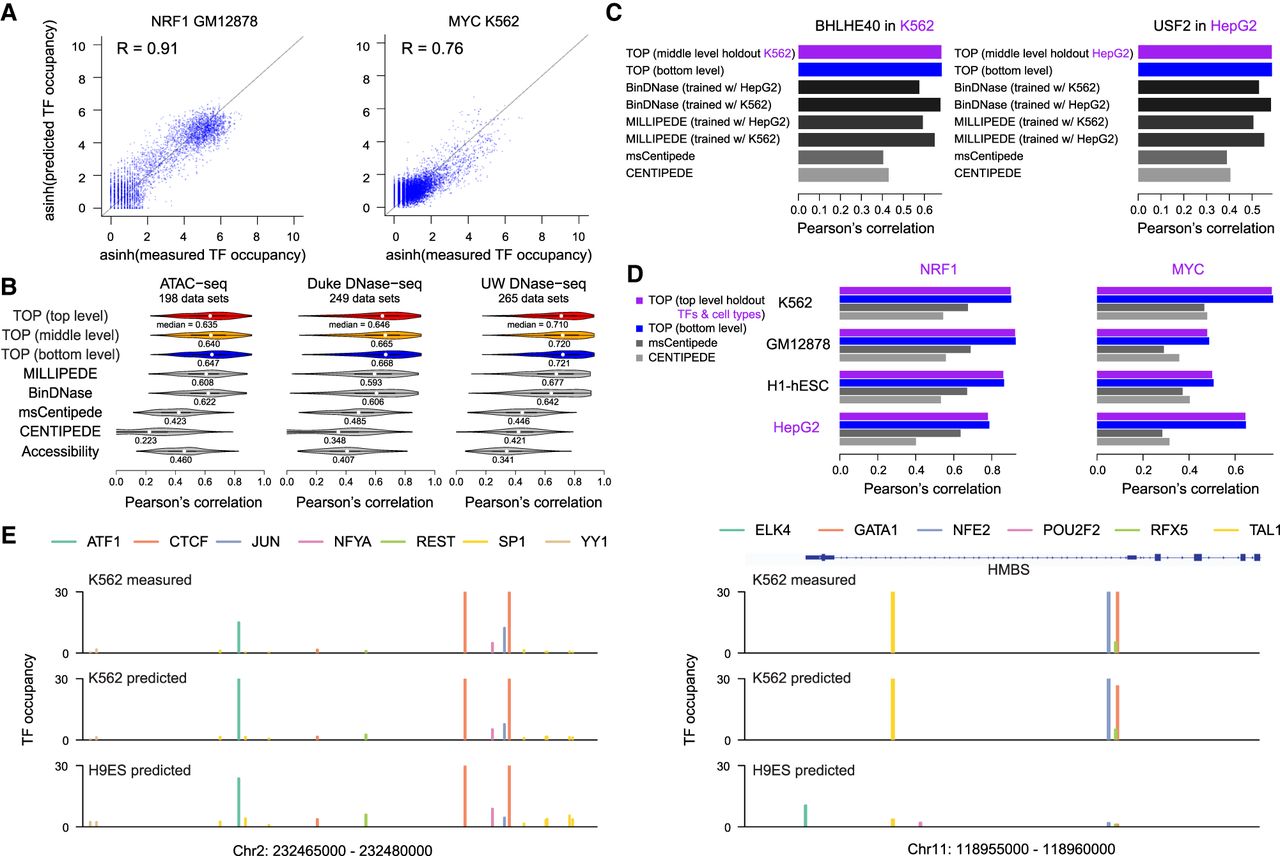
Evaluation of TOP results. (A) Scatter plots show predicted versus measured TF occupancy for test chromosomes of a specific TF in a specific cell type using Duke DNase-seq data, with dots representing the candidate binding sites across the genome. Model performance differs among TFs, as seen in the two examples. (B) Separately for ATAC-seq and DNase-seq data from Duke and UW protocols, violin plots show distribution of Pearson's correlations between predicted and measured TF occupancy (asinh transformed) in test chromosomes. Predictions were made with TOP models at each level of the hierarchy, in comparison with CENTIPEDE, msCentipede, MILLIPEDE, and BinDNase (using log odds of binding probability as a quantitative measurement of TF occupancy), as well as with total accessibility around candidate sites. (C) Comparing prediction performance in scenarios in which ChIP training data are missing for a cell type (indicated in purple). (Left) Predicting TF occupancy with data from K562 held out from training. (Right) Predicting TF occupancy with data from HepG2 held out from training. TOP is trained without any held-out data. MILLIPEDE and BinDNase are trained using a different cell type and also using data that was held out for TOP, showing that TOP performs as well without the held-out data as these methods do with it. msCentipede and CENTIPEDE are unsupervised, so do not require training data; however, their performance is poor. For more, see Supplemental Figure S5. (D) Comparing prediction performance in scenarios in which ChIP training data are missing for both TFs and a cell type (indicated in purple). Data from NRF1 and MYC (and all their TF family members with similar motifs), as well as from HepG2, were held out from training. MILLIPEDE and BinDNase were not included in this comparison as they require training data from the exact TFs. For more, see Supplemental Figure S6. (E) Predicted TF occupancy landscapes for two genomic regions in K562 and H9ES cell types. For K562, ChIP-seq data for these TFs are available and are displayed for comparison; for H9ES, no published ChIP-seq data are available so TOP provides a novel view of TF occupancy in this embryonic stem cell line. (Left) An example genomic region where the occupancy landscape did not change markedly between K562 and H9ES. (Right) An example genomic region near the HMBS gene (involved in heme biosynthesis) where GATA1, TAL1, and NFE2 showed clear cell type–specific occupancy.








