
Accumulation and ineffective silencing of transposable elements on an avian W Chromosome
Abstract
One of the defining features of transposable elements (TEs) is their ability to move to new locations in the host genome. To minimize the potentially deleterious effects of de novo TE insertions, hosts have evolved several mechanisms to control TE activity, including recombination-mediated removal and epigenetic silencing; however, increasing evidence suggests that silencing of TEs is often incomplete. The crow family experienced a recent radiation of LTR retrotransposons (LTRs), offering an opportunity to gain insight into the regulatory control of young, potentially still active TEs. We quantified the abundance of TE-derived transcripts across several tissues in 15 Eurasian crows (Corvus (corone) spp.) raised under common garden conditions and find evidence for ineffective TE suppression on the female-specific W Chromosome. Using RNA-seq data, we show that ∼9.5% of all transcribed TEs had considerably greater (average, 16-fold) transcript abundance in female crows and that >85% of these female-biased TEs originated on the W Chromosome. After accounting for differences in TE density among chromosomal classes, W-linked TEs were significantly more highly expressed than TEs residing on other chromosomes, consistent with ineffective silencing on the former. Together, our results suggest that the crow W Chromosome acts as a source of transcriptionally active TEs, with possible negative fitness consequences for female birds analogous to Drosophila (an X/Y system), in which overexpression of Y-linked TEs is associated with male-specific aging and fitness loss (“toxic Y”).
Transposable elements (TEs) are mobile genetic elements that are characterized by their ability to move to new genomic locations (McClintock 1948, 1950). As sources of molecular variation, TEs can have beneficial effects on organismal fitness (Trizzino et al. 2017; Schrader and Schmitz 2019; Rutter et al. 2020; Sundaram and Wysocka 2020). However, TE insertions can also disrupt gene function or initiate chromosomal rearrangements, thereby reducing host fitness. In humans, for instance, more than 120 diseases are caused by TE insertions (Hancks and Kazazian 2016). Hosts can reduce the impact of deleterious TEs through removal or silencing. Removal of TEs by purifying selection is less effective when recombination rates are low, as tends to be the case in sex-limited chromosomes (Y or W) (Dolgin and Charlesworth 2008). Removal efficacy is additionally affected by the effective population size of the target, which is highest in autosomes, intermediate in sex chromosomes with at least one copy in both sexes (X or Z), and lowest in the sex-limited chromosomes (Charlesworth and Charlesworth 1983; Charlesworth and Langley 1989). An alternative way of limiting the potentially negative effects of TEs is to suppress their activity (Deniz et al. 2019). In vertebrates, this primarily involves chemical modifications at TE loci (“epigenetic silencing”) such as cytosine methylation (Deniz et al. 2019).
TE activity was long thought to be restricted to the early developing germline, where epigenetic repression is relaxed (Zamudio and Bourc'his 2010). However, in several taxa, ubiquitous TE activity has been shown to occur in healthy adult tissue, suggesting that TE silencing is often incomplete (Esteve-Codina et al. 2011; Dong et al. 2017; Brunet et al. 2018). It has been suggested that incomplete TE silencing may reflect a pervasive trade-off between epigenetic TE silencing and host genome function, as the former can interfere with the expression of nearby genes (for review, see Choi and Lee 2020). If occurring on sex-limited chromosomes, such trade-offs have important consequences for the heterogametic sex. For example, in Drosophila (an X/Y system), incomplete suppression of TEs on the Y Chromosome is thought to increase the mutational burden in males (“toxic” Y) (Wei et al. 2020), potentially contributing to male-specific ageing (Brown et al. 2020). In X/Y systems, TEs on the sex-limited chromosome thus determine general sex-specific differences. In female-heterogametic Z/W systems, such as birds, the sex-limited W Chromosome often shows exceptionally high TE densities (Smeds et al. 2015; Warren et al. 2017; Bertocchi et al. 2018; Peona et al. 2021). It is conceivable that W-linked TEs have broad sex-specific (fitness) effects in Z/W systems analogous to X/Y systems. However, whether the avian W Chromosome acts as a toxic reservoir of transcriptionally active TE copies remains poorly understood, with only one recent study reporting evidence for expression of female-specific, likely W-linked, endogenous retroviruses (ERVs) in gonadal tissue of the emu, chicken, and zebra finch (Peona et al. 2021).
Avian genomes harbor comparatively few TEs (∼4%–10%, compared with up to 60% in other tetrapod vertebrates) (Zhang et al. 2014; Gao et al. 2017; Sotero-Caio et al. 2017). However, the large clade of passerine birds appears to have experienced a recent diversification of LTR retrotransposons (Kapusta and Suh 2017; Suh et al. 2018). Two observations point to a particularly high activity of LTRs in the crow family. First, in a genome-wide comparison of 48 bird species, the American crow (Corvus brachyrhynchos) had more than twice as many copies of ERVs (an LTR superfamily) than all the other species (1032 compared with an average of 335) (Cui et al. 2014). Second, an in-depth annotation of the first version of the hooded crow (Corvus (corone) cornix) assembly revealed a high diversity of lineage-specific, evolutionarily young LTRs in this species (Vijay et al. 2016). Young TEs are particularly likely to still be intact and therefore capable of transposition, offering the possibility to gain insight into their regulatory control.
In this study, we exploit the high contiguity of the current hooded crow genome and added newly assembled W chromosomal sequence to quantify the genomic abundance and transcription of TEs in 15 Eurasian crows raised under common garden conditions.
Results
W Chromosomal sequence
To identify W Chromosome–linked sequences in the hooded crow genome, we generated a de novo genome assembly for a female individual using Oxford Nanopore Technologies (ONT) long-read sequencing (1.12 Gb total size and 9.3 Mb contig N50 after filtering; see Methods). We then combined a sex-specific coverage-based approach with synteny information from the W Chromosome of the closely related New Caledonian crow (Corvus moneduloides) to assign to the W Chromosome. The resulting 101 contigs had a total sequence length of 12.3 Mb and were ordered by synteny to the New Caledonian crow W Chromosome (21.5 Mb), the only other corvid W Chromosome to date. This W assembly was added to the hooded crow reference genome (GCA_000738735.5) and was used for all subsequent analyses.
Recent expansion of LTR retrotransposons on the W
RepeatMasker identified a total of 189,169 distinct repeat sequences (Supplemental Table S1). Of these, 187,409 were bona fide TEs (Class I or Class II), with the remainder consisting of simple repeats, low-complexity repeats, satellite DNA, rRNA, and tRNA (Table 1). Repeat content, expressed as the proportion of genomic bases belonging to repeat sequence, varied among chromosome types and was lowest on autosomal sequence (∼6.5%), intermediate on the Z Chromosome (∼11.9%), and highest by far on the W Chromosome (∼84.8%). Genome-wide, the vast majority of TE copies were LINE elements (72.4%), followed by LTR retrotransposons (14.6%), DNA transposons (6.5%), and SINE elements (3.3%) (Table 1; Supplemental Table S1), which is consistent with previous findings in other avian taxa (Warren et al. 2017; Kapusta and Suh 2017; Suh et al. 2018). LTR retrotransposons occupied a similar amount of genomic sequence as LINE elements on the autosomes and the Z Chromosome but were by far the most dominant type of TE on the W Chromosome (Fig. 1A; Supplemental Fig. S1).

Abundance and transcription of TEs annotated in the hooded crow reference genome shown by chromosomal class and by TE type (LTRs, blue; LINEs, orange; SINEs, yellow; DNA transposons, gray; other TEs, light purple). (A) TE content expressed as the proportion of TE sequence relative to total sequence length. (B) Proportion of transcribed TEs relative to the total number of TE copies. (A) Autosomes; (Z) Z Chromosome; (W) W Chromosome.
Summary of the genomic abundance and transcription of annotated repeats by repeat type
Transcription of LINES and young LTR retrotransposons
Of the 187,409 bona fide TE copies annotated in the crow reference genome, 13,940 (7.4%) were transcribed in our population sample as per our definition (0.5 or more transcripts per million in at least four of the 15 samples) (Supplemental Table S2). This constitutes more than half of all transcribed genomic features (26,714) in our data set. The proportions of autosomal, Z chromosomal, and W chromosomal TEs that were transcribed were ∼6.3%, 6.9%, and 23.4%, respectively (Fig. 1B). Despite its relatively small size, the W Chromosome not only was therefore the most TE-rich (Fig. 1A) but also had the highest proportion of transcriptionally active TEs (Fig. 1B). On the autosomes and the Z Chromosome, LINEs were the most frequently expressed TE type; in contrast, on the W Chromosome, LTRs were both the most abundant and the most frequently expressed TE type (Supplemental Fig. S2). A number of autosomes also contained regions with a high density of transcribed TEs (e.g., Chromosome 13) (Supplemental Fig. S2); however, these were often located near subtelomeric regions, and the maximum density of transcribed TEs on any autosome was lower than the average density of transcribed TEs on the W Chromosome.
Assigning all annotated TE copies to bins of increasing divergence from their respective, family-level, consensus sequences (Kapusta et al. 2017) shows that, across all chromosome types, a large proportion of LTRs was young (0%–1% divergence) (Fig. 2A), whereas LINEs were predominantly older (>10% divergence) (Fig. 2A). On all three chromosome types, older LTRs (>10% divergence) were rarely transcribed, whereas transcription of LINEs appeared to have no such age restriction (Fig. 2B).
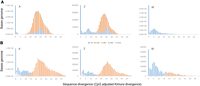
Frequency distribution of TE sequence in bins of increasing divergence from the consensus sequence shown separately for autosomes (left), the Z Chromosome (middle), and W Chromosome (right). (A) Histogram of all annotated TEs; (B) all TE copies that show evidence for transcription in our population sample. Note the shift in abundance versus transcription for LTRs.
Influence of tissue and sex on TE expression
We used Tau as an indicator of expression specificity (Yanai et al. 2005). The Tau index ranges between zero and one, where zero indicates broad expression, and one indicates expression specific to one tissue (Kryuchkova-Mostacci and Robinson-Rechavi 2017). Of all 13,940 transcribed TEs, 6641 were expressed only in one tissue (Tau = 1). A further 2192 TEs were highly tissue specific (Tau ≧ 0.8) but were expressed in more than one tissue. Instances of TEs with Tau values ≧ 0.8 were recorded 487 times in liver, 3625 in spleen, 2459 in testis, and 2276 in ovary (Supplemental Table S3).
To decompose the variance of transcript abundance across tissues, we performed principal component analysis (PCA) on the normalized and variance-stabilized counts of reads mapped to annotated genes and TEs, respectively. As expected, PCA of genes showed a clear partitioning of variance by tissue, with the first two PC axes accounting for 92% of the variation (Fig. 3A). Except for gonadal tissue (ovary, testis), sex contributed only marginally to the overall variation in genes (Fig. 3A; Supplemental Fig. S3). In contrast, TE transcript counts showed a clear partitioning of variance among the sexes for all tissues, with the first two PC axes explaining 72% of the variation (Fig. 3B). This is consistent with a signal not only of tissue specificity but also of sex-specificity. Neither genes nor TEs showed any clear separation of corone and cornix subspecies (for taxonomic considerations, see Supplemental Text).

Principal component analysis of RNA-seq data. PC1 and PC2 of normalized and vst-transformed read counts for coding genes (A) and TEs (B) in the hooded crow genome (including W-linked contigs).
To further explore the extent of taxon and sex bias in TE expression, we performed a differential expression (DE) analysis using DESeq2 with a linear model including terms for subspecies, tissue, and sex. The repetitive nature of TE elements means that an unknown number of short reads may map to multiple locations in the genome, thereby biasing estimates of TE transcript abundance. Using only uniquely mapping reads increases the rate of true positives but underestimates the signal of transcription associated with younger TE families (Teissandier et al. 2019). One solution is to include multimapping reads in analyses of TE expression but to randomly report only one position (Teissandier et al. 2019). To guard against misinterpretation of the data owing to the choice of one or the other mapping method, we used both (see Methods).
In both random and unique mapping mode, only six TEs (four CR1 LINEs and two LTRs) were differentially expressed between subspecies (Supplemental Table S4). This observation is consistent with the observed lack of separation by subspecies in our PCA as well as previous gene expression studies in this system (Poelstra et al. 2014, 2015).
Contrary to taxon, sex had a strong effect on TE expression. Almost 10% of all 13,940 transcribed TEs showed DE between the sexes (random mode, 1328; unique mode, 1247) (Supplemental Table S5). Nearly all of these differentially expressed TEs (DETEs) showed higher transcript abundance in females compared with males (random mode, 1285 or 96.8%; unique mode, 1193 or 95.6%). The vast majority of female-biased DETEs originated on the W Chromosome (unique mode, 1135 or 91.0%) or had a copy on the W Chromosome indistinguishable from copies elsewhere in the genome (random mode, 1136 or 88.4%) (Fig. 4A,B). Autosomes and the Z Chromosome also contributed to female-biased TE expression, but to a much lower extent: In random mode, 146 DETEs mapped to the autosomes and 46 to the Z Chromosome (Fig. 4A; Supplemental Table S5). In unique mode, the numbers were similar, with 110 DETEs mapping to the autosomes and 51 to the Z Chromosome (Fig. 4B; Supplemental Table S5). Even in unique mode, a handful of male DNA-seq reads mapped to the W Chromosome (Fig. 4B; Supplemental Table S5). This likely reflects the presence of TE copies in the male genome that are highly similar to one or more W-linked TEs.
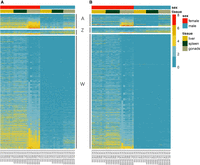
Normalized and log-transformed RNA-seq read counts of autosomal, Z chromosomal, and W chromosomal DETEs across three different tissues in females versus males using the “random” (A) and “unique” (B) mapping approaches, respectively. Note that even in unique mode, some of the W-linked DETEs attracted male RNA-seq reads. This observation is consistent with some of the W-based TEs having highly similar copies elsewhere in the genome.
Ineffective silencing of W-linked TEs
We were interested to assess whether overexpression of W-linked TEs was independent of both ploidy (only one copy of the Z in females; no copy of the W in males) and TE abundance (highest relative TE abundance on the W Chromosome). We tested the effect of ploidy on TE expression using a linear model, in which we asked whether TE transcript abundance (RNA-seq) was higher in females than in males after accounting for TE abundance (DNA-seq) (Supplemental Methods). We found that TE expression (RNA-seq counts) was significantly higher in females than in males across all chromosomal classes and tissues, including from TEs residing on the Z Chromosome (Supplemental Fig. S4). This suggests that TE transcript abundance is generally higher in females and that TE copies on the W Chromosome are driving this pattern.
Next, we asked whether TE transcription from the W Chromosome was elevated above and beyond what is expected based on the high abundance, particularly of (young) LTRs, on the W Chromosome compared with autosomes and the Z Chromosome (Figs. 1B, 2B). To examine this question, we limited our considerations to females, for which direct comparisons of abundance versus expression are possible for all three chromosomal classes (A, Z, W) (Supplemental Methods). Specifically, we asked whether transcript levels from W-linked TEs exceeded expectations based on their abundance on this chromosome. We conducted this analysis separately for LTR elements, of which only young copies were expressed (Fig. 2B), and for the substantially older LINE elements. We found evidence for up-regulation of W chromosomal, but not autosomal or Z chromosomal, TEs of both types in the liver and spleen. In gonads, in which sexual selection, particularly on the Z Chromosome, is expected to confound general patterns, this effect was not observed (Supplemental Methods). Overall, these results suggest that female genomes, and in particular the W Chromosome, constitute a permissive environment for TE transcription.
Mixed effect of TEs on the expression of neighboring genes
TE insertions can disrupt coding sequence or modify the transcriptional activity of neighboring genes. To assess whether TE insertions in or near coding sequences are selected against, we first determined the number of TEs that reside within 10 kb of annotated protein coding genes. Of the 187,409 bona fide TEs in the hooded crow genome, 370 (0.2%) overlapped with sequence of annotated genes (Supplemental Table S6), and a further 31,789 (17.0%) were found within 10 kb upstream of or downstream from a gene. Of the 370 genes containing TEs, 38 contained more than one (up to six) TE(s), though some of those instances might represent nested TEs rather than individual copies.
To assess if the vicinity of TEs influenced gene expression, we compared the normalized read counts of genes containing TE sequence 0–2 kb, 2–10 kb, and >20 kb away from transcription units. There was no statistically significant difference in expression levels of genes with TEs in their immediate vicinity (0–2 kb; median = 0.05, N = 10,788) and genes with TEs >20 kb away (median = 0.09, N = 888; Mann–Whitney U test, P > 0.05, two-tailed). In contrast, genes with TEs located 2–10 kb away (N = 13,568) had significantly lower expression levels (median = 0.02) than genes in the other two distance bins (Mann–Whitney U test, P < 0.0001, two-tailed).
Next, we assessed whether sex bias in TE expression might be driven by sex-biased expression of nearby genes. For example, the autosomal and Z chromosomal components of female bias in TE expression may either be owing to ineffective TE silencing in females or result from coexpression between TEs and neighboring, female-biased genes. Of the sex-specific DETEs identified in unique or random mode, respectively, only three were located within the transcription unit of genes (Supplemental Table S7), and none of them were differentially expressed between males and females. A further 44 genes were located within 10 kb of a sex-biased DETE (Supplemental Table S7); of these, only one was itself differentially expressed between the sexes (C17orf58 homolog). Assuming equal power for inferring sex bias in protein coding genes and TEs, we infer that sex bias in TE expression is independent of sex bias in protein coding genes in our study system.
Discussion
In this study, we characterized the genomic and transcriptomic repertoire of repetitive elements in a population sample of an avian lineage experiencing recent TE expansion. Inferences are based on short-read sequencing data from DNA and RNA mapped to a reference genome. This approach comes with the limitation that many TEs segregating at low frequencies in single or few individuals will go undetected (Weissensteiner et al. 2020). Despite this limitation, our work revealed strong tissue, sex, and chromosome effects on TE expression.
Repeat content
We report an average repeat content of ∼6.5% for the autosomes of the hooded crow genome. This figure falls toward the higher end of the range observed in birds (∼4%–10%) (Zhang et al. 2014) but corresponds closely with the repeat content of the American crow (C. brachyrhynchos) genome (7.4%) (Zhang et al. 2014). Consistent with theoretical predictions (Charlesworth 1991), and as shown for sex-limited chromosomes in other taxa (Śliwińska et al. 2016), TE content was highest (84.8%) on the W Chromosome. Higher tolerance of TE insertions on the W likely results from a combination of reduced effective population size, absence of recombination, and low gene density (3/6 genes per megabase on the W vs. 12/14 on the Z, and an average of 26/39 on autosomes in Corvus (corone) spp./C. moneduloides) (Weissensteiner et al. 2020). Smeds et al. (2015) reported a TE content of ∼48.5% across 6.9 Mb of the nonrecombining part of the collared flycatcher W Chromosome, a short-read based assembly. Repetitive DNA, including TEs, are often underrepresented in short-read assemblies (Peona et al. 2018). Higher levels of missing data in the short-read-based flycatcher genome compared with the long-read-based crow genome examined here could explain the higher TE content found in crows; however, species-specific differences in the density of TEs on avian W Chromosomes could also explain the difference: In six recently analyzed, reference-quality, avian W assemblies, TE densities ranged between 22% and 80% (Kapusta and Suh 2017; Peona et al. 2021).
A comparison of the dominant TE classes on the different chromosomes revealed an overabundance of CR1 LINE elements on the autosomes and the Z Chromosome, and of LTR retrotransposons on the W Chromosome, a pattern previously also seen in flycatchers (Suh et al. 2018). The observation that ∼85% of the crow W Chromosome consisted of TEs and that most of the W-based TEs were young LTR retrotransposons lends indirect support to the hypothesis that the avian W Chromosome may act as a refugium for transcriptionally active TEs (Smeds et al. 2015; Warren et al. 2017; Suh et al. 2018; Peona et al. 2021).
TE distribution
In the hooded crow genome, only ∼0.2% of TEs reside within transcriptional units of coding genes. This figure increases to ∼20% if we include sequence 10 kb upstream of and downstream from coding genes. Overall, these figures correspond closely with those reported for other avian genomes. For example, in the zebra finch, 16% of all TEs are reported to reside within 10 kb of either end of transcription units; similarly, in the chicken genome, ∼25% of endogenous retroviruses were either within transcriptional units or within 10 kb of each end (Bolisetty et al. 2012).
TE activity
Uncontrolled TE activity can interrupt functional sequence and result in deleterious, large-scale genome rearrangements. TE activity in healthy organisms has therefore long been assumed to be restricted to the early developing germline, embryonic tissue, and the placenta (Slotkin and Martienssen 2007). However, it is becoming increasingly clear that TE regulation is often incomplete, with important implications for host genome stability and fitness (Hollister and Gaut 2009; Brown et al. 2020; Wei et al. 2020). Using a third-generation reference genome and two different pipelines to quantify TE transcript abundance, we show that ∼10% of the annotated TEs in the crow genome escape silencing.
The successful transposition of TEs depends on the full transcription of a number of TE-associated genes. The transposition of LTR retrotransposons, in particular, requires a full-length, sense-strand transcript, including the proteins encoded by the gag and pol genes. Over time, mutations in the host genome will lead to a deterioration of TE sequence and a corresponding decrease in activity. Transcribed LTR retrotransposons in our system fall predominantly into young age bins (<8% divergence from the consensus sequence), supporting the expectation whereby transcribed TEs are mostly young. CR1 LINEs did not follow this pattern, showing evidence for transcription of far older elements instead.
TE expression was highly tissue specific, corroborating findings from studies in other vertebrates. In humans, for instance, multiple studies have reported tissue-specific expression of TEs, with a recent study showing that TE expression was as predictive of tissue groupings as gene expression (Chung et al. 2019). Similarly, in rats, separation by tissue/organ explained ∼95% of the variance in TE expression (Dong et al. 2017). In birds, tissue-specific TE expression has also been reported for the chicken (Bolisetty et al. 2012). In a study of the human TE transcriptome, tissue-specific TE expression covaried with the expression of nearby genes and was owing to the presence on TEs of binding sites for transcription factors that regulates expression in a given tissue (Trizzino et al. 2017). As we found no evidence for strong patterns of gene–TE coexpression in our study system, tissue-specific TE expression may best be explained by trans-acting regulatory mechanisms.
A substantial number of TEs were expressed in the gonadal tissue of both sexes, consistent with TEs being generally expressed in gonads (Dechaud et al. 2019). Gonadal tissue contains germline cells at various stages of their development, including the earliest stages, when epigenetic repression is relaxed (Zamudio and Bourc'his 2010). In this study, ovarian tissue had among the highest level of transcription, mirroring results in an XY system (rats), in which testis tissue had among the highest levels of TE expression (Dong et al. 2017).
Sex-biased TE expression
Approximately 10% of transcribed TEs were differentially expressed between the sexes. Recent studies of TE expression in mammals (Dong et al. 2017; Trizzino et al. 2017) and birds (Peona et al. 2021) have also reported sex differences in TE expression. In contrast to our finding, however, neither study observed a pattern of consistent overexpression in only one of the sexes across several tissues, and the number of expressed TEs with a detectable sex bias was small. In rats, for example, only 26 TEs were found to be differentially expressed between the sexes, compared with more than 1000 in this study. As in our study, most (84.6%) of the TEs showing sex-biased expression in rats were LTR retrotransposons. Because of the way in which they propagate, LTR retrotransposons are thought to have a greater potential for deleterious effects on host fitness than other TE classes (Faulkner et al. 2009). Elevated activity of W-based LTRs in female crows might therefore increase the mutational load and negatively affect female fitness analogous to the negative fitness effects of Y-linked TEs observed in Drosophila males (Brown et al. 2020).
High transcript abundance of TEs in females may simply reflect excess abundance of young, transcriptionally active TE copies on the female W Chromosome. However, we found TE transcription to be elevated even when controlling for TE abundance and TE age effects: The probability of a TE being expressed is elevated across the entire genome in females and particularly so on the W Chromosome. This finding suggests trans-acting effects on TE expression that are likely driven by the W Chromosome. An analogous pattern of male-biased expression of Y-linked TEs has recently been described in two Drosophila species, Drosophila pseudoobscura and Drosophila miranda. As in our study, the observed sex bias in TE expression was largely owing to elevated expression of Y-linked TEs in the heterogametic sex (males in Drosophila; females in crows) (Wei et al. 2020), and although it was confined to early developmental stages in D. pseudoobscura, it persisted into adulthood in D. miranda. In Drosophila, the availability of a well-annotated Y Chromosome proved essential in identifying the conflict between transcription of functional Y-linked genes and suppression of Y-linked TEs as a potential reason for the observed incomplete silencing of Y-linked TEs: euchromatin, required for transcription, antagonizes the formation of heterochromatin, a major mechanism of TE silencing. The avian W Chromosome contains dozens of functional genes (Xu and Zhou 2020). It is therefore conceivable that a similar mechanism might explain the incomplete silencing of W-based TEs in female birds. Our study contributes to accumulating evidence that sex-limited chromosomes in general, and the W Chromosome in particular, may have roles beyond sex determination and gonadal development and that Y-/W-linked repeats, and polymorphisms thereof, can have genome-wide epistatic effects (Lemos et al. 2010; Kutch and Fedorka 2017).
The trans-regulating capabilities of TEs are known from several systems, in which TE-derived RNAs regulate either distant genes or the activity of TEs themselves (Piergentili 2010; Kawaoka et al. 2011; McCue and Slotkin 2012; Wang et al. 2017; Cho 2018). Trans-regulatory activity of W-based TEs in the crow would explain our observation of a female bias in TE expression generally, including hundreds of autosomal and even Z chromosomal TEs. This result may thus be a first indication of a tentative regulatory role of W-linked repeats in birds.
Overall, our work provides evidence for a female heterogametic analogue of the toxic Y in X/Y systems with important implications for our understanding of transcriptional control of TEs, TE-induced fitness effects, and long-term TE propagation.
Methodological considerations
For most protein-coding genes, each individual contains at least a single gene copy. In the absence of mRNA molecule counts, we can conclude with some confidence that this gene is not expressed. More generally, we expect a direct relationship between mRNA abundance and transcription activity (except for a minority of recently duplicated genes). In contrast, TEs segregate in a population with strongly skewed site frequency spectra. As a consequence, the majority of TE copies are unique to a single or few individuals. Only a small minority of copies will be shared between all individuals (Weissensteiner et al. 2020). As a consequence, the lack of an mRNA signal may result either from lack of expression or from the absence of a syntenic copy in the reference genome. Using a single genome as mapping reference does not allow us to distinguish between these possibilities. Moreover, RNA-seq reads from a copy that is absent in the reference genome runs an increased risk of multimapping to similar copies elsewhere in the genome.
This population genetic reality has consequences for the interpretation of our results. First, the reference genome will only represent a fraction of the TE inserts present in the population sample. Our estimate that 7.4% of all TEs are transcribed therefore likely constitutes an underestimate. Second, multimapping will be increased beyond levels already prevalent with short-read sequencing data. This expectation motivated us to use different mapping approaches (unique vs. random mode, see Methods) to assess the robustness of the results. Furthermore, statistical inference of factors modulating transcription activity required the inclusion of a DNA-seq reference experiencing the same reference bias. Third, results exploring the effect of TEs on expression of nearby genes are to be interpreted with caution.
To circumvent the above-mentioned reference bias for insertion–deletion polymorphisms, future studies of TE activity will require a data set combining transcriptome data with haplotype-resolved de novo assemblies for all individuals under investigation. Both data types would strongly benefit from the use of long-read data to assure reliable inference of structural variation (Tusso et al. 2019) and minimize the effect of multimapping. This “pangenome” approach has become a reality for the study of TEs in small eukaryotic genomes (Tusso et al. 2022) and is starting to be within reach for vertebrate-size genomes (Weissensteiner et al. 2020; De Coster et al. 2021).
Methods
Sampling and data generation
In May 2014, crow hatchlings of an approximate age of 21 d were obtained directly from the nest using an unmanned aerial vehicle to assess nest status (Weissensteiner et al. 2015). Hooded crows (C. (corone) cornix) were sampled in the area around Uppsala, Sweden (59°52′N, 17°38′E); carrion crows (C. (corone) corone) in the area around Konstanz, Germany (47°45N′, 9°10′E) (Supplemental Table S7). To avoid any confounding effects of relatedness, only a single individual was selected from each nest. After transfer of carrion crows to Sweden by airplane, all crows were hand-raised indoors at the Tovetorp field station, Sweden (58°56′55′′N, 17°8′49′′E). When starting to feed by themselves. they were released to large roofed outdoor enclosures (6.5 × 4.8 × 3.5 m) specifically constructed for the purpose. All crows were maintained under common garden conditions in groups of a maximum of six individuals separated by subspecies and sex. In October 2016, at an age of ∼2.5 yr, individuals were euthanized by cervical dislocation. Tissues were immediately harvested and stored at −80°C until extraction.
Regierungspräsidium Freiburg granted permission for the sampling of wild carrion crows in Germany (Aktenzeichen 55-8852.15/05). Import into Sweden was registered with the Veterinäramt Konstanz (Bescheinigungsnummer INTRA.DE.2014.0047502) and its Swedish counterpart Jordbruksverket (Diarienummer 6.6.18-3037/14). Sampling permission in Sweden was granted by Naturvårdsverket (Dnr: NV-03432-14) and Jordbruksverket (Diarienummer 27-14). Animal husbandry and experimentation was authorized by Jordbruksverket (Diarienummer 5.2.18-3065/13, Diarienummer 27-14) and ethically approved under the directive 2010/63/EU on the Protection of Animals used for Scientific Purposes by the European Research Council (ERCStG-336536).
DNA and RNA extraction and sequencing
RNA was extracted from the liver, spleen, and gonadal tissue of seven C. (c.) corone (N = 4 males, and N = 3 females) and eight C. (c.) cornix (N = 3 males, and N = 5 females), respectively, using the RNeasy plus universal extraction kit (Qiagen) and following the manufacturer's guidelines. Libraries were prepared from 500 ng total RNA using the TruSeq stranded mRNA library preparation kit (Illumina), which includes removal of ribosomal RNA using poly(A) selection. Sequencing of 50-bp single-end reads was performed by the SNP&SEQ Technology Platform at Uppsala University, Sweden, using Illumina HiSeq 2500 v. 2 chemistry and a target sequencing depth of 20×.
High-molecular-weight DNA was extracted from a female C. (c.) cornix individual (NCBI BioSample [https://www.ncbi.nlm.nih.gov/biosample] SAMN13509843), sampled from the same subpopulation as the (male) individual that was used for the reference genome. Four flow cells of the ONT minION sequencer yielded 29.55 Gb of raw data (read N50: 13.47 kb), and one flow cell of the ONT promethION sequencer yielded 38.67 Gb of raw data (read N50: 13.11 kb).
Identification and assembly of W-linked contigs
We then assembled individual long sequence reads with the flye assembly algorithm (Kolmogorov et al. 2019). The initial whole-genome assembly consisted of 8875 contigs with an average size of 128 kb (median, 3.2 kb) and a contig N50 of 9.21 Mb. For further downstream analysis, we restricted the minimum contig length to 10 kb, leading to an exclusion of 6778 contigs and a total assembly size of 1.117 Gb. We then used two complementary approaches to identify W-linked contigs: a synteny-based approach and a coverage-based approach. For the synteny-based approach, we chose the Vertebrate Genomes Project's (VGP) assembly of the New Caledonian crow (C. moneduloides), which includes among the best W Chromosome assemblies available for birds from the corvid family. We downloaded the C. moneduloides primary assembly from the VGP website (https://vgp.github.io/genomeark/Corvus_moneduloides/, accessed 15 November 2020) and aligned all contigs > 10 kb (2197 out of 8875) against the C. moneduloides W Chromosome using LASTZ (Harris 2007). This approach yielded 227 potentially W-linked contigs.
The coverage-based approach relies on the fact that the W Chromosome is female-limited. Sequencing reads from male crows should therefore not map against W chromosomal sequence. To identify potential W-linked contigs, we mapped both Illumina short reads and Pacific Biosciences (PacBio) long reads of a male C. (c.) cornix from the Swedish population (BioSample SAMN02439830, accession number SRS602284) to the 8875 contigs of our assembly using NGMLR (Sedlazeck et al. 2018). We limited the maximum number of segments per kilobase of reference a read can align to 3 (‐‐max_segments = 3). We used mosdepth (Pedersen and Quinlan 2018) to calculate coverage for each contig. Excluding contigs with a mean coverage < 3 and retaining only contigs > 50 kb yielded 302 potentially W-linked contigs. After excluding all contigs that were identified by only one of the two approaches, our W assembly consisted of 101 contigs with a combined sequence of 12.27 Mb. Comparing these 101 with the previously identified pseudoautosomal region (PAR) in the crow genome yielded no conclusive linear alignments (Catalán et al. 2021). We therefore conclude that the PAR is absent in our set of W-linked sequences. To further illustrate that the 101 contigs are of W Chromosome origin, we mapped both the female long-read sequencing data described above and Illumina short-read sequencing data of four more hooded crows (two males and two females; NCBI Sequence Read Archive [SRA; https://www.ncbi.nlm.nih.gov/sra] accession numbers ERR2900302, ERR2900303, ERR2900305, ERR2900306) to the hooded crow reference assembly and the W-linked contigs identified in this study. We then calculated read depth in 10,000 randomly subsampled 10-kb windows on Chromosome 1 and compared it to the same number of subsampled windows on W-linked contigs, the Z Chromosome, and the autosomes. Supplemental Figure S5 shows the relative read depths of all chromosomes and shows that the W-linked contigs meet the expectation of half of autosome coverage in females and approaching zero in male individuals.
TE content
Pipelines for the discovery and annotation of TEs were run on the combined sequence of the (male) crow reference genome (NCBI Assembly [https://www.ncbi.nlm.nih.gov/assembly] accession number: GCA_000738735.5) and the newly generated assembly of W-linked contigs. We used three different pipelines for the discovery and annotation of TEs: RepeatMasker (https://www.repeatmasker.org), CARP (Zeng et al. 2018), and RetroTector (Sperber et al. 2007). CARP uses pairwise alignment and single linkage clustering to identify families of repeats, with a bias toward low-divergence, potentially still active, TEs (Zeng et al. 2018). RetroTector specializes in the identification and characterization of retroviral sequences and is potentially also biased toward the detection of evolutionarily younger elements (Sperber et al. 2007). Both RetroTector and CARP were run using default settings. Following the investigators’ recommendations, we used a score cut-off of 300 for the retroviral sequences identified by RetroTector. To annotate the (evolutionarily young) repeats identified by CARP, we used zebra finch consensus sequences available on Repbase (https://www.girinst.org/repbase/index.html; accessed April 9, 2018), consensus sequences of TEs identified in the flycatcher genome (Suh et al. 2018), and consensus sequences of TEs identified in an earlier version of the hooded crow genome (Vijay et al. 2016). We then ran RepeatMasker on the combined genomic sequence using our TE library together with the inbuilt chicken database and under default settings.
TE age estimates
We used the degree of sequence divergence from the consensus sequence as a rough estimate of the age of TEs. Sequence divergence of TEs from their consensus was computed from the RepeatMasker output files (.align) following Kapusta et al. (2017) and using the Perl script “parseRM.pl” (https://github.com/4ureliek/Parsing-RepeatMasker-Outputs/blob/master/parseRM.pl). Estimates of sequence divergence obtained using this approach are sufficiently accurate for our purpose, which is to compare the broad age ranges of LINEs and LTRs across the different chromosomal classes.
Quantification of TE-derived transcripts
Following removal of Illumina adaptors and reads with a Phred quality score below 20 in Trim Galore v. 0.4.4 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), RNA-seq reads were mapped against the merged sequence of the hooded crow reference genome and the W-linked contigs using STAR v2.5.2b (Dobin et al. 2013). We followed recent recommendations by Teissandier et al. (2019) for the study of transposon expression and regulation (see also Supplemental Text, section 2). Specifically, we quantified TE transcript abundance using the “random” counting method, in which each multimapping read contributes one randomly chosen valid alignment (Teissandier et al. 2019). To assess how including versus excluding multimapping reads affected the conclusions drawn from our analyses, we compared this approach with one that only considers uniquely mapping reads (“unique” mode) (but see Supplemental Text, section 2). For read mapping, we adjusted the flags in STAR as follows: ‐‐outFilterMultimapNmax 1000 ‐‐outSAMmultNmax 1 ‐‐outFilterMismatchNmax 3 ‐‐outMultimapperOrder Random ‐‐winAnchorMultimapNmax 1000 ‐‐alignEndsType EndToEnd ‐‐alignIntronMax 1 ‐‐alignMatesGapMax 350. We used the featureCounts function of the R (R Core Team 2017) package Subread (Liao et al. 2019) for read counting, setting countMultiMappingReads to true in random mode and to false in unique mode. For mapping summary statistics, see Supplemental Table S8.
Counts were normalized using the median of ratios normalization (MRN) implemented in the R package DESeq2 (Love et al. 2014). We considered TEs to be transcribed if transcript abundance exceeded 0.5 transcripts per million in at least four samples. Tests for DE were performed following the standard DE analysis steps of DESeq2 and using the design formula ∼tissue + subspecies + sex. Differentially expressed genomic features were identified at a log2 fold change (LFC) threshold greater than one, corresponding to a greater than twofold expression difference between test groups and a false-discovery rate (FDR) ≤10%.
Quantification of TE abundance in the crow genome
We were interested to assess the genomic abundance of individual TEs to relate genomic TE abundance to abundance of transcribed TE copies. The number of normalized DNA-seq reads mapping to a particular TE is expected to be roughly proportional to the number of times this TE appears in the DNA sample, thus reflecting its genomic abundance (Magi et al. 2012). To compare RNA- and DNA-seq read abundance for the same TE, we normalized counts from both types of data using median of ratios method (MRN). Although principally used for RNA-seq data, the MRN has recently been shown to be suitable for the normalization of DNA-seq data for the purpose of gene abundance estimation (Pereira et al. 2018).
In the absence of resequencing data for the 15 individuals used for the RNA-seq study, we used Illumina resequencing data of 10 individuals (N = 5 females and N = 5 males) from an Italian crow population (NCBI SRA accession number PRJEB9057) (Supplemental Table S7). Assuming an excess of low-frequency TE insertions segregating in the population (Bourgeois and Boissinot 2019), not all TEs identified in the reference genome will be present in nonreference individuals. As such, we will likely underestimate absolute abundances. However, because site-frequency spectra of crows sampled across Europe are near identical (Vijay et al. 2016), the same bias will apply to all samples, therefore allowing relative comparisons between DNA-seq (Italian samples) and RNA-seq data (German, Swedish samples).
DNA-seq reads, initially 100 bp long, were trimmed to a final length of 50 bp, the same length as the RNA-seq reads. Reads were then mapped against our crow assembly (including the W-linked contigs) using NextGenMap v.0.5.4 (Sedlazeck et al. 2013) with a minimum identity i = 0.88, corresponding to the minimum identity used for the RNA-seq data, and default values otherwise. Reads were counted using the function featureCounts in the R package subReads with countMultiMappingReads set to false. Raw DNA-seq read counts were normalized using MRN. Although principally used for RNA-seq data, the median of ratios method has recently been shown to be suitable for the normalization of DNA-seq data for the purpose of gene abundance estimation (Pereira et al. 2018).
Comparison of TE abundance and transcription
For each individual crow, read counts (DNA-seq for Italian crows, RNA-seq for Swedish/German crows) were averaged across the DETEs identified in unique mode. A linear model was fit to the log-transformed counts using sex (male or female), tissue (liver, spleen or gonad), and nucleic acid type (RNA or DNA) as categorical explanatory variables (formula: log(counts) ∼ sex + tissue + type + sex × type). To test whether transcription of W-linked TEs remains higher than that of autosomal and Z-linked TEs after accounting for differences in TE abundance, we limited our considerations to the female data set, for which a direct comparison of abundance versus expression is possible between all three chromosomal classes (A, Z, W). We asked whether TE transcript levels (RNA-seq) correspond to TE abundance (DNA-seq) across all chromosome classes or whether transcripts from W-linked TEs are elevated (statistical interaction RNA-seq/DNA-seq × A/Z/W). To account for the higher relative abundance specifically of (young) LTR elements on the W Chromosome, we conducted this analysis separately for LTR elements, of which only young copies were expressed, and for the substantially older LINE elements. For each tissue separately, we then fit a linear model to the female data using the formula: log(counts) ∼ type + chromosomal class + type × chromosomal class. All analyses were performed in R version 4.0.0 (R Core Team 2017).
Distance to transcription units
The distance of TEs from the nearest annotated transcription unit (coding sequence, 3′ UTRs, and 5′ UTRs) was determined using the function closest in BEDTools v2.29.2 (Quinlan and Hall 2010), and the data were cataloged in 2-kb intervals. A more detailed binning of TE locations with respect to different classes of coding sequence was not possible owing to the preliminary state of the annotation for the crow genome version used here.
Data access
All raw sequencing data generated in this study and the newly assembled W-linked sequence have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA772570. All custom scripts used for the analysis of the data presented here are available from GitHub (https://github.com/EvoBioWolf/2022_Warmuth_GenomeRes) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Sven Jakobsson for providing the infrastructure for animal husbandry at Tovetorp research station. We also thank Christen Bossu for her contribution in obtaining samples. Thomas Giegold, Nils Andbjer, Tamara Volkmer, Barbara Martin Schitsch, Luisa Sontheimer, and Joanna Schinner were of invaluable support in raising and maintaining the captive crow population. Martin Wikelski, Inge Müller, and additional staff from the Max-Planck-Institute for Ornithology in Radolfzell facilitated sampling in Germany and transport to Sweden. We further thank Dirk Metzler for advice on statistical analyses, Saurabh Pophaly for assistance with the MYSQL database required for RetroTector, and Justin Meröndun for producing Supplemental Figure S5. We acknowledge funding from European Research Council (ERCStG-336536 to J.B.W.W.) and LMU Munich (to J.B.W.W.) and financial contribution from Stockholm University to Tovetorp field station.
Author contributions: V.M.W. and J.B.W.W. conceived of the study and wrote the paper with input from M.H.W. V.M.W. conducted all analyses except for the W assembly, which was performed by M.H.W. J.B.W.W. and M.H.W. obtained crow hatchlings in the field; J.B.W.W. was responsible for the common garden experiment and sampled the tissue material.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275465.121.
- Received March 4, 2021.
- Accepted February 8, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








