
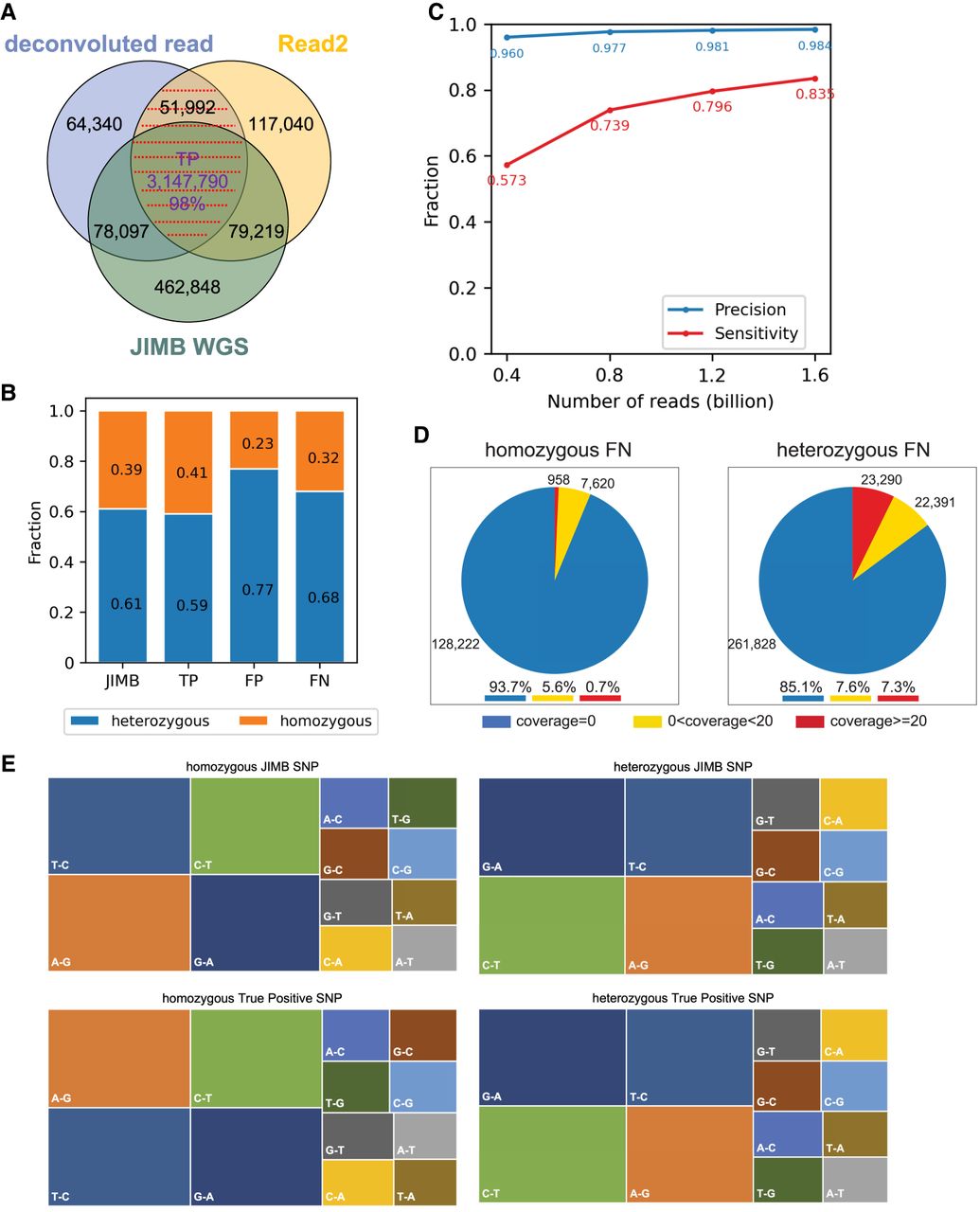
SNP identification. (A) Comparison of SNPs identified using Methyl-SNP-seq deconvoluted read and Read2 with those using JIMB whole-genome sequencing data. Common SNPs, which were identified by both deconvoluted read and Read2 and marked by red dashed lines, are referred to as Methyl-SNP-seq defined SNPs. (B) Fraction of heterozygous and homozygous Methyl-SNP-seq defined SNPs. (C) Precision and sensitivity of SNP identification using different numbers of Methyl-SNP-seq reads. Precision = TP/(TP + FP). Sensitivity = TP/(TP + FN) with TP: True positive. FP: False positive. FN: False negative. (D) Distribution of the genome coverage of the false-negative SNP sites. (E) Characterization of the JIMB and true-positive Methyl-SNP-seq defined SNPs (i.e., T-C means in the VCF file REF = T while ALT = C). The JIMB whole-genome sequencing of NA12878 was used as a benchmark for comparison.








