
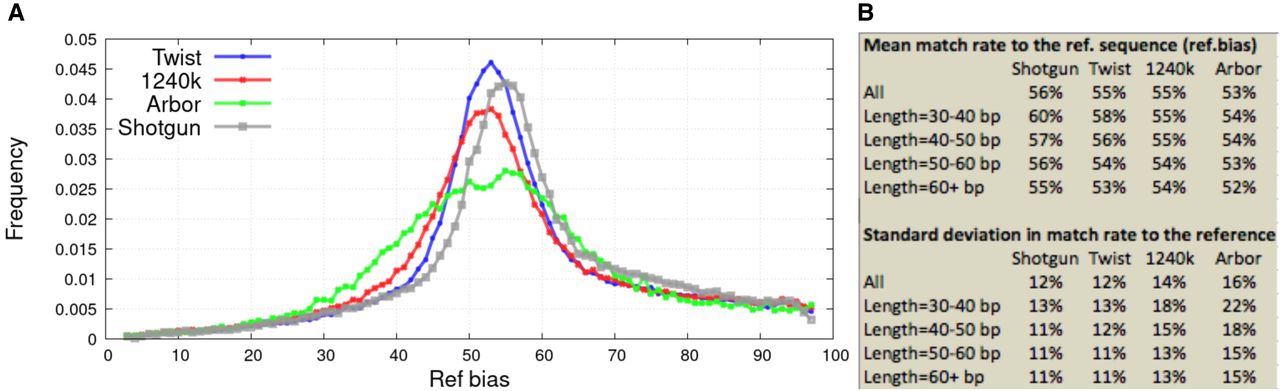
Figure 4.
Variation in reference bias across SNPs. (A) All analyses are based on sequences from loci ascertained as highly likely to be heterozygous, corrected for stochastic error in the estimates using the expectation maximization (EM) algorithm described in Supplemental Text S2. (B) Mean and standard deviation of EM-corrected distributions stratified by sequence length (longer sequences align more reliably so have less bias). Results for this figure reflect data before removal of duplicated sequences.








