
Abstract
Topologically associating domains (TADs) were recently identified as fundamental units of three-dimensional eukaryotic genomic organization, although our knowledge of the influence of TADs on genome evolution remains preliminary. To study the molecular evolution of TADs in Drosophila species, we constructed a new reference-grade genome assembly and accompanying high-resolution TAD map for D. pseudoobscura. Comparison of D. pseudoobscura and D. melanogaster, which are separated by ∼49 million years of divergence, showed that ∼30%–40% of their genomes retain conserved TADs. Comparative genomic analysis of 17 Drosophila species revealed that chromosomal rearrangement breakpoints are enriched at TAD boundaries but depleted within TADs. Additionally, genes within conserved TADs show lower expression divergence than those located in nonconserved TADs. Furthermore, we found that a substantial proportion of long genes (>50 kbp) in D. melanogaster (42%) and D. pseudoobscura (26%) constitute their own TADs, implying transcript structure may be one of the deterministic factors for TAD formation. By using structural variants (SVs) identified from 14 D. melanogaster strains, its three closest sibling species from the D. simulans species complex, and two obscura clade species, we uncovered evidence of selection acting on SVs at TAD boundaries, but with the nature of selection differing between SV types. Deletions are depleted at TAD boundaries in both divergent and polymorphic SVs, suggesting purifying selection, whereas divergent tandem duplications are enriched at TAD boundaries relative to polymorphism, suggesting they are adaptive. Our findings highlight how important TADs are in shaping the acquisition and retention of structural mutations that fundamentally alter genome organization.
Higher-order folding of eukaryotic genomes in the nucleus partitions the genome into multiple spatial layers, ranging from DNA loops to chromatin domains to compartments (Rowley and Corces 2018). A growing consensus has recognized chromatin domains, often referred to as topological associating domains (TADs), as fundamental units of three-dimensional (3D) genome organization (Szabo et al. 2018). Such domains are closely linked to important DNA-dependent and cellular processes, such as DNA replication (Pope et al. 2014), transcription (Schoenfelder and Fraser 2019), DNA-damage repair (Schmitt et al. 2016), development, and cell differentiation (Zheng and Xie 2019). The presence of TADs or TAD-like domains has been widely characterized in species as varied as yeast (Mizuguchi et al. 2014), bacteria (Le et al. 2013), plants (Liu et al. 2017), and animals (Fishman et al. 2019), suggesting they represent a conserved feature of genome organization (Szabo et al. 2019).
TADs are thought to serve as regulatory units for controlling gene expression by promoting and constraining long-range enhancer–promoter interactions (Schoenfelder and Fraser 2019). Genes localizing within the same TAD tend to be coregulated and coexpressed (Nora et al. 2012). Additionally, changes in TADs or their boundaries can alter expression of genes, including developmental and disease-related genes (Lupiáñez et al. 2015; Bonev et al. 2017; Akdemir et al. 2020). However, recent work re-examined the tight relationship between gene regulation and TADs by observing that disruption of TAD features can alter expression for only a small number of genes (Despang et al. 2019; Ghavi-Helm et al. 2019). Evolutionary comparisons across species (Krefting et al. 2018; Eres et al. 2019) permit us to quantify the nature of functional constraint in these domains, potentially reconciling the disparate observations above.
The conservation of TADs between distantly related species is associated with the preservation of synteny across vast spans of evolutionary time (Dixon et al. 2012). Genome rearrangement breakpoints are more common at TAD boundaries than inside TADs, implying that a subset of TADs is constrained to remain intact through evolution (Krefting et al. 2018; Lazar et al. 2018). In addition, deletions are depleted at TAD boundaries (Huynh and Hormozdiari 2019; Sadowski et al. 2019; Akdemir et al. 2020), presumably because deleting boundaries might destroy the insulating effect separating neighboring TADs. Thus, TAD boundaries share hallmarks of other functional genomic regions, like strong evolutionary constraints (Fudenberg and Pollard 2019). However, other than the dearth of deletions at TAD boundaries, it remains unclear whether natural selection drives the acquisition and fate of other classes of mutations, like duplications and transposable element (TE) insertions, when they occur at TAD boundaries.
TADs have been extensively analyzed using Hi-C in embryos (Sexton et al. 2012) and cell lines in Drosophila melanogaster (Li et al. 2015; Wang et al. 2018; Chathoth and Zabet 2019). Hi-C data from other Drosophila species are primarily targeted for genome scaffolding and are too sparse in coverage for high-resolution spatial organization analysis (Bracewell et al. 2019). In this study, we introduced a new genome assembly and created a high-resolution Hi-C contact map for Drosophila pseudoobscura, which diverged from D. melanogaster ∼49 million years ago (Thomas and Hahn 2017). We assessed 3D genome conservation between D. pseudoobscura and D. melanogaster using TADs annotated with high-coverage Hi-C data. We also investigated the association between TADs and gene expression by analyzing public RNA-seq data set (Yang et al. 2018). Additionally, we characterized genome rearrangement breakpoints in 17 Drosophila species (Mahajan et al. 2018; Miller et al. 2018) and their distribution along TAD regions. Finally, we investigated patterns of structural variants (SVs) at TAD boundaries with SV genotypes derived from reference-quality assemblies for intraspecies (14 D. melanogaster strains) (Chakraborty et al. 2019) and interspecies (D. melanogaster vs. three simulans clade species; D. pseudoobscura vs. D. miranda) (Mahajan et al. 2018; Chakraborty et al. 2020) comparisons. This study provides accurate, high-resolution resources for Drosophila genome structure research, and our findings highlight the evolutionary significance of TADs in shaping genome rearrangements, SVs, and gene expression.
Results
A new reference genome assembly for D. pseudoobscura
We de novo assembled the genome of D. pseudoobscura females from the strain MV-25-SWS-2005 using deep (280×) coverage Pacific Biosciences (PacBio) long reads (Supplemental Table S1). The resulting assembly consists of 72 contigs and is extremely contiguous, accurate, and complete (assembly size, ∼163.3 Mb; N50 contig length, 30.7 Mb; base-pair accuracy QV score, 52; and BUSCO [Simão et al. 2015], 99.6%) (Supplemental Tables S2, S3). The vast majority of three telocentric autosomes (Chr 2, Chr 3, and Chr 4) and the dot chromosome, together with the complete circular mitochondrial DNA genome (mtDNA) are each assembled into a single contig. The X Chromosome is represented in three contigs, including both arms of the euchromatic region and a repeat-rich contig (∼9.7 Mb) showing enrichment of centromere-specific repeats at both ends. These three contigs were combined into a single scaffold based on Hi-C contact maps (Supplemental Fig. S1). Additionally, we recovered 64 small and repetitive contigs (totaling ∼6.6 Mb) that were not scaffolded (Supplemental Table S4). The final assembly spans the majority of every major chromosome, interrupted only by two sequence gaps near the pericentromeric region of the X Chromosome (Supplemental Fig. S2). Although the middles of the chromosome arms are assembled contiguously, there are likely gaps remaining in the repetitive sequences at the ends of chromosomes, particularly in the centromeric and pericentromeric heterochromatin.
The current assembly is the most contiguous genome assembly for D. pseudoobscura (Supplemental Table S5). Approximately 26.9% (43.9 Mb) of the genome is annotated as repeat sequences, including 13.95% (22.77 Mb) derived from retrotransposons (Supplemental Table S6). We annotated 13,413 gene models using RNA-seq and full-length mRNA sequencing (Iso-Seq) data (Supplemental Tables S6, S7). The largest structural difference between our assembly and two other assemblies from different strains (English et al. 2012; Bracewell et al. 2019) is an X-linked pericentromeric inversion (∼9.7 Mb). We verified this inversion with Hi-C contact frequency data, and our sequenced strain shares a configuration with a closely related species, Drosophila miranda (Supplemental Fig. S2; Mahajan et al. 2018). The distribution of genomic features is generally conserved between D. pseudoobscura and D. miranda, despite several large chromosomal rearrangements reshuffling their genomes (Supplemental Fig. S2; Bartolomé and Charlesworth 2006). We identified 1.43 million SNPs (about 10.3 per kbp), 0.53 million indels (<50 bp; about 3.7 per kbp), and 8227 SVs (≥50 bp) that affected ∼1.8 Mb genomic regions between our assembly and the Dpse4.0 assembly from the reference strain (Supplemental Fig. S3; English et al. 2012).
Identification of TADs in D. pseudoobscura adult full bodies
We generated 397 million Hi-C paired-end reads (2 × 150 bp) from cross-linked DNA extracted from full bodies of adult females. We used the method of Arima Genomics that uses multiple restriction enzyme cutting sites for chromatin digestion, leading to a theoretical mean restriction fragment resolution of ∼160 bp. After filtering, about half of the raw data are retained for construction of the Hi-C contact map (Supplemental Table S8). This subset of read pairs yields a contact map with a maximal estimated “map resolution” of ∼800 bp, as calculated following the approach of Rao et al. (2014).
TAD annotation may vary moderately among different computational methods (Forcato et al. 2017). To examine the consistency of TAD calls across methods, we identified TADs at 5-kbp resolution using three tools: HiCExplorer (Ramírez et al. 2018), Armatus (Filippova et al. 2014), and Arrowhead (Fig. 1A; Durand et al. 2016), which inferred 1013, 3352, and 795 TADs, respectively. After excluding TADs <30 kbp, Armatus retained 858 TADs (mean length, 123 kbp), Arrowhead retained 795 (mean length, 140 kbp), and HiCExplorer retained 996 (mean length, 146 kbp) (Fig. 1B,C). These tools yield results with different properties. For example, Arrowhead allows nested TADs and permits them to be spaced discretely, resulting in adjacent TADs separated by gaps. On the other hand, most adjacent TADs among HiCExplorer calls share boundaries. Partly for this reason, the calls from alternative methods do not overlap completely. Despite this, 589 TADs are shared by at least two tools, covering 57% (92.5/163 Mb) of the genome (Fig. 1B).
TADs annotated using full body samples in D. pseudoobscura. (A) Hi-C contact map (5-kbp resolution) on a ∼3-Mb region from the X Chromosome with TADs annotated using Arrowhead (black), Armatus (green), and HiCExplorer (blue). The bottom browser tracks show the local profiles of binding sites of BEAF-32 and CTCF, as well as two histone marks (H3K4me3 and H3H27me3). (B) Overlap of TADs and their genome coverage annotated using three tools. (C) TAD size distribution for three tools. Vertical dashed lines represent the mean values. (D) Enrichment of CTCF and BEAF-32 binding sites at HiCExplorer TAD boundaries (P < 1.0 × 10−4). (E) Enrichment of ATAC-seq signal (open chromatin marks) at HiCExplorer TAD boundaries (P < 1.0 × 10−4). (F) TAD boundaries (HiCExplorer) are enriched in H3K4me3 (P < 1.0 × 10−4) but depleted for H3K27me3 marks (P < 1.0 × 10−4). P-values (D–F) were determined by permutation tests (n = 10,000); dashed lines represent mean values obtained from permutation tests; gray shaded areas, mean ± SD in D and 95% intervals in E and F.
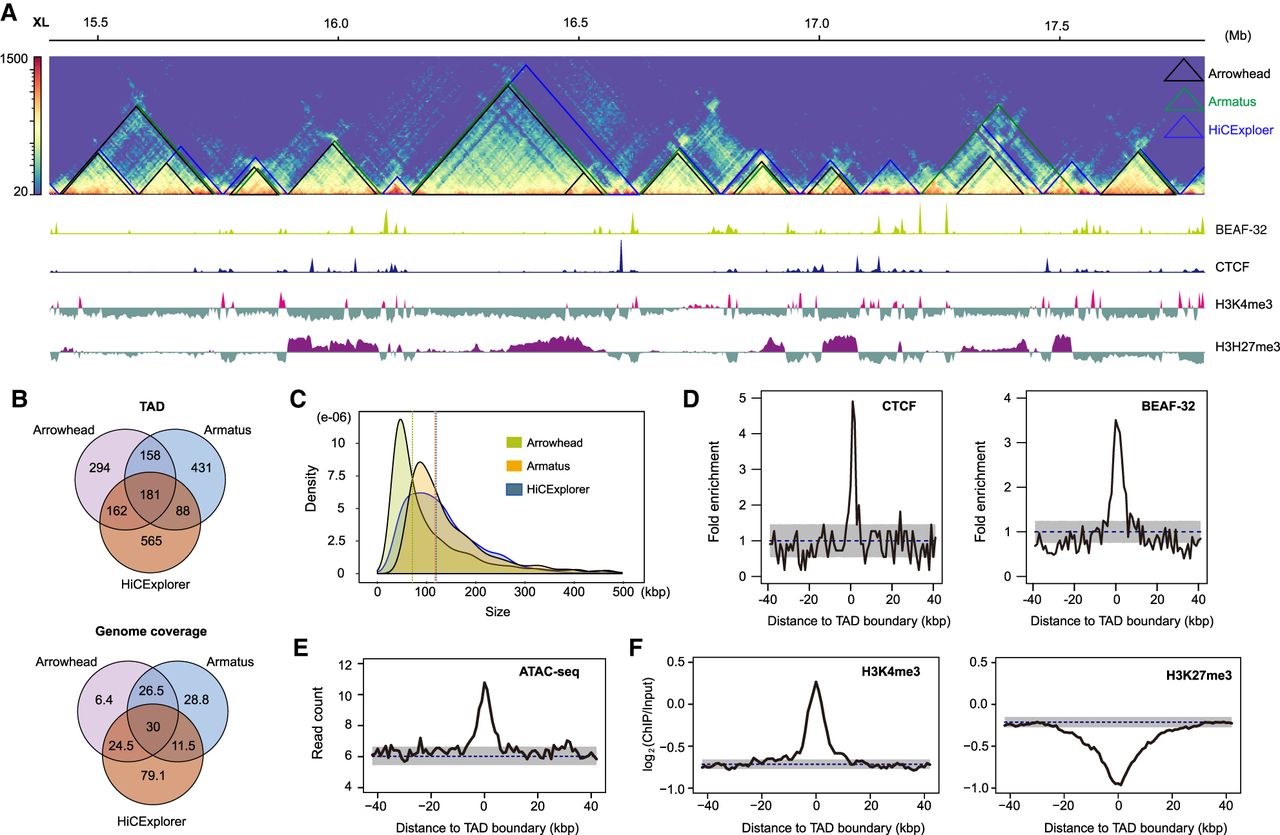
To determine whether the TADs we inferred were supported by biological features, we investigated the chromatin landscape surrounding their boundaries using publicly available data (Ni et al. 2012; Yang et al. 2012; Schuettengruber et al. 2014). We found that TAD boundaries from all three tools are enriched for CTCF and BEAF-32 binding sites (Fig. 1D; Supplemental Fig. S4), as well as open and active chromatin as inferred from ATAC-seq data and H3K4me3 marks, respectively (Fig. 1E,F; Supplemental Fig. S4), but are depleted for repressive chromatin marks, H3K27me3 (Fig. 1F; Supplemental Fig. S4). These observations are consistent with earlier studies in D. melanogaster (Hug et al. 2017; Wang et al. 2018; Chathoth and Zabet 2019), suggesting that Hi-C data from full bodies identifies a considerable amount of biologically meaningful TAD features in Drosophila. As HiCExplorer TAD calls are most strongly correlated with biological features (Fig. 1D–F; Supplemental Fig. S4), we used this set of TADs in most of the subsequent analysis, unless otherwise noted.
Evolutionary conservation of TADs between D. pseudoobscura and D. melanogaster
We investigated the TAD conservation between two distantly related Drosophila species, D. pseudoobscura and D. melanogaster. We first identified blocks of synteny between their genomes. The resulting synteny map consists of 985 orthologous blocks >10 kbp, with an average size of 101 kbp in D. melanogaster and 109 kbp in D. pseudoobscura, spanning 74.6% (100/134 Mb) of the D. melanogaster genome and 70% (110/157 Mb) of the D. pseudoobscura genome, respectively (Supplemental Table S9). The high-quality genome assemblies increased the average length of synteny blocks by 20% (100–83/83 kbp) compared to an earlier study (Richards et al. 2005). The synteny map (Fig. 2A) reveals extensive genome shuffling between species, but most of the orthologous blocks are located in the same Muller elements, suggesting that even on a small scale, translocations rarely occur between chromosomes in the course of Drosophila genome evolution, consistent with previous observations (Muller 1940; Schaeffer 2018).
Evolutionary conservation of TADs between D. pseudoobscura (Dpse) and D. melanogaster (Dmel). (A) Genome synteny map between Dmel and Dpse constructed using 985 syntenic blocks >10 kbp. (B) Conservation of TADs on a ∼1.2-Mb orthologous region between Dmel and Dpse. (C) Overlap of TAD features across three D. melanogaster cell lines. (D) Upset plot showing the overlap of TAD boundaries across three D. melanogaster cell lines and D. pseudoobscura whole body; 5-kbp boundaries for Dmel and 10-kbp boundaries for Dpse. (E) Upset plot showing genome coverage that maintained conserved TADs across three D. melanogaster cell lines and D. pseudoobscura. (F) Distribution of size variation (Dmel size divided by Dpse size) of orthologous TADs between Dmel and Dpse. (G) Correlation of the size of conserved TADs between Dmel and Dpse. (H) Correlation between the size difference of the orthologous TADs and the size difference of local synteny blocks where the orthologous TADs are located between Dmel and Dpse. (Dif.) Difference.
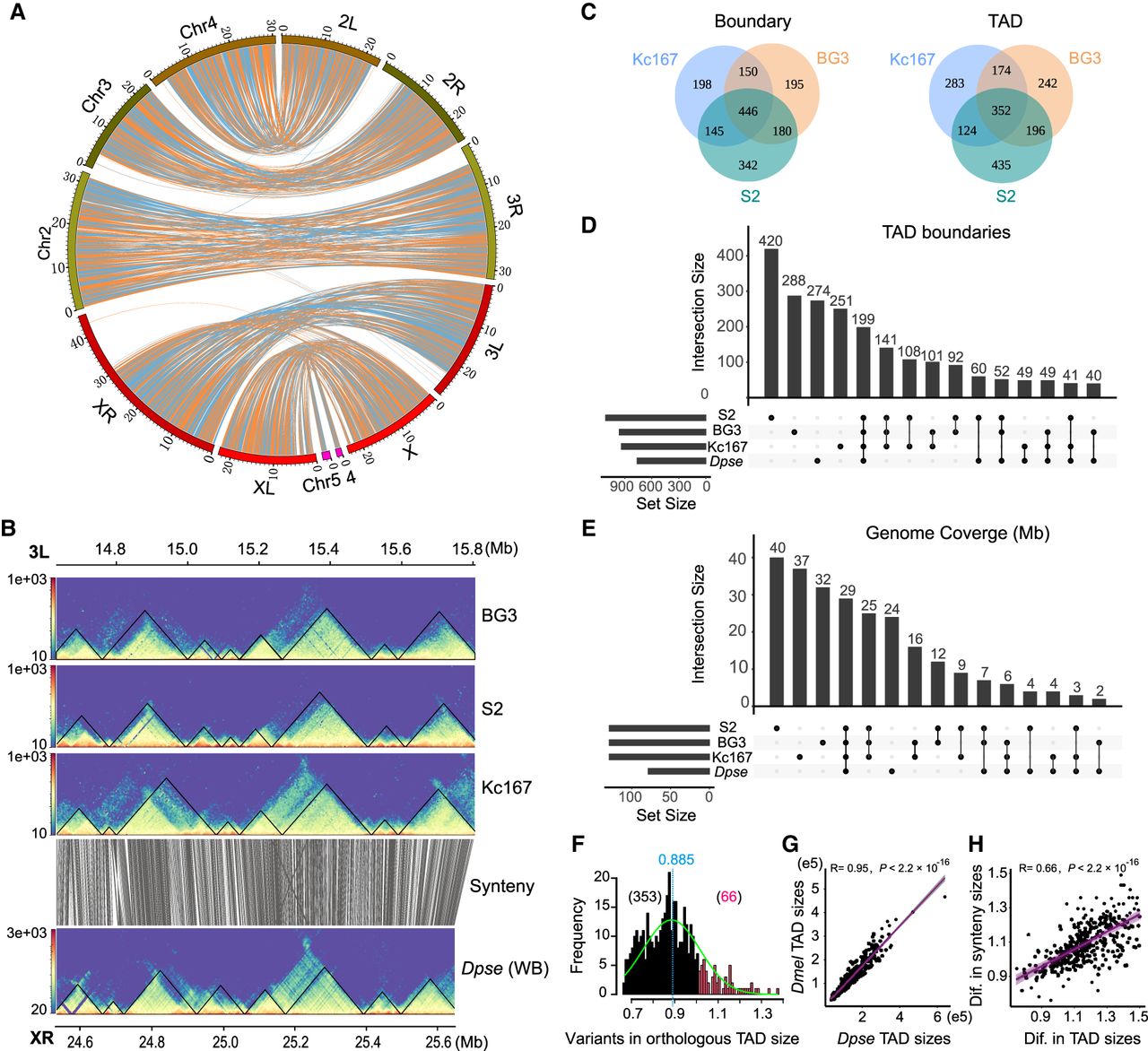
For D. melanogaster, we annotated TADs using published Hi-C data from three cell lines (S2, Kc167, and BG3) (Supplemental Table S10; Wang et al. 2018; Chathoth and Zabet 2019). TADs were annotated using HiCExplorer as described for our D. pseudoobscura data. Consistent with previous studies (Ulianov et al. 2016), TADs and their boundaries are largely shared across cell lines (Fig. 2B; Supplemental Figs. S5, S6). About 68% of TADs and 76% of their boundaries (10-kbp boundaries) are shared at least in two cell lines, whereas 32% and 24% of the TADs and boundaries are cell line specific (Fig. 2C).
For each interspecific comparison, genomic coordinates of TAD features (i.e., body and boundary) were lifted over between species (Table 1). The success rate for each lift is 66.9%–71.5% for bodies and 72.5%–75.4% for boundaries, comparable to their shared genome synteny fraction (70%–74.6%). Of TADs annotated across the three D. melanogaster cell lines, we found that 29.4%–32% of body annotations and 31.5%–35.3% of boundary annotations were conserved with D. pseudoobscura (Table 1). The conserved TADs cover at least 34.5% (46.2/134 Mb) of the D. melanogaster genome. In the reciprocal comparison, ∼41.4% of TADs, spanning ∼39.7% (62.4/157 Mb) of the D. pseudoobscura genome, and 48.5% of the boundaries were conserved with D. melanogaster (Table 1). Such rates of conservation are substantially higher than the background levels observed from permuted genomic coordinates based on two different null hypotheses (Table 1; Methods). We note that adjusting the stringency of the reciprocal overlap requirement from 80% to 90% did not qualitatively alter the results but decreased the conservation rate (e.g., genome coverage) from 39.7% to 27.8% (Supplemental Table S12). Thus, the conservative estimate is that nearly half of the syntenic regions of the genome (30%–40% of the total genome) retained conserved TADs between D. melanogaster and D. pseudoobscura (Fig. 2D,E; Supplemental Fig. S7), and TADs in the remaining half may have diverged. This estimate is consistent with our visual inspection of Hi-C contact maps across the long syntenic regions between these two species (Fig. 2B; Supplemental Fig. S5). The inferred conservation magnitude is comparable to some regulatory phenotypes between these two species, such as functional enhancer conservation (46%) (Arnold et al. 2014) and CTCF binding sites (30%) (Ni et al. 2012), but lower than others, such as BEAF-32 (>70%) (Yang et al. 2012) and alternative splicing (∼80%) (Malko 2006).
Summary of TAD annotation (HiCExplorer), lift, and conservation between D. melanogaster (Release 6) and D. pseudoobscura (this study)

Three hundred fifty-three out of 419 conserved TADs are larger in D. pseudoobscura than their orthologs in D. melanogaster, whereas the rest are larger in D. melanogaster (Fig. 2F). The sizes of the orthologous TADs are correlated with the sizes of the local syntenic blocks (Fig. 2G,H), suggesting that TADs may expand or contract proportionally with their local genomic regions. On average, D. pseudoobscura TADs are ∼10% larger than their counterparts in D. melanogaster, smaller than the ∼17% difference (157–134/134 Mb) observed between their genome sizes, suggesting that size variation among homologous TADs is more strongly constrained than genome size variation elsewhere.
Conservation of different TAD boundary classes
TAD boundaries may differ in insulation strengths, binding affinity of insulators, occurrence across cell types, and the flanking chromatin states. These properties may distinguish their evolutionary conservation. To test this, we first assessed the conservation of boundaries overlapping with binding sites for six insulator proteins in each of the three D. melanogaster cell lines (Kc167, BG3, and S2) (Supplemental Table S13). We found that TAD boundaries overlapping BEAF-32, CP190, and Chromator binding sites are more frequently shared between species than those lacking these sites. However, we observed no such pattern for CTCF, Su(Hw), and Trl (Fig. 3A; Supplemental Fig. S8). These findings are consistent with previous observations that BEAF-32, CP190, and Chromator are better predictors of TAD boundaries in Drosophila (Ramírez et al. 2018; Wang et al. 2018).
Conservation of distinct classes of TAD boundaries between D. melanogaster and D. pseudoobscura. (A) Boundaries that overlap with binding sites of architectural proteins versus those lacking the corresponding binding sites. (B) Boundaries linked with active TADs versus inactive TADs. (C) Boundaries that share across cell lines versus cell-line-specific boundaries. (D) Strong versus weak boundaries. Statistical significance were calculated using chi-square test: (***) P < 0.001; (**) P < 0.01; (NS) no significance (for details, see Supplemental Table S14).
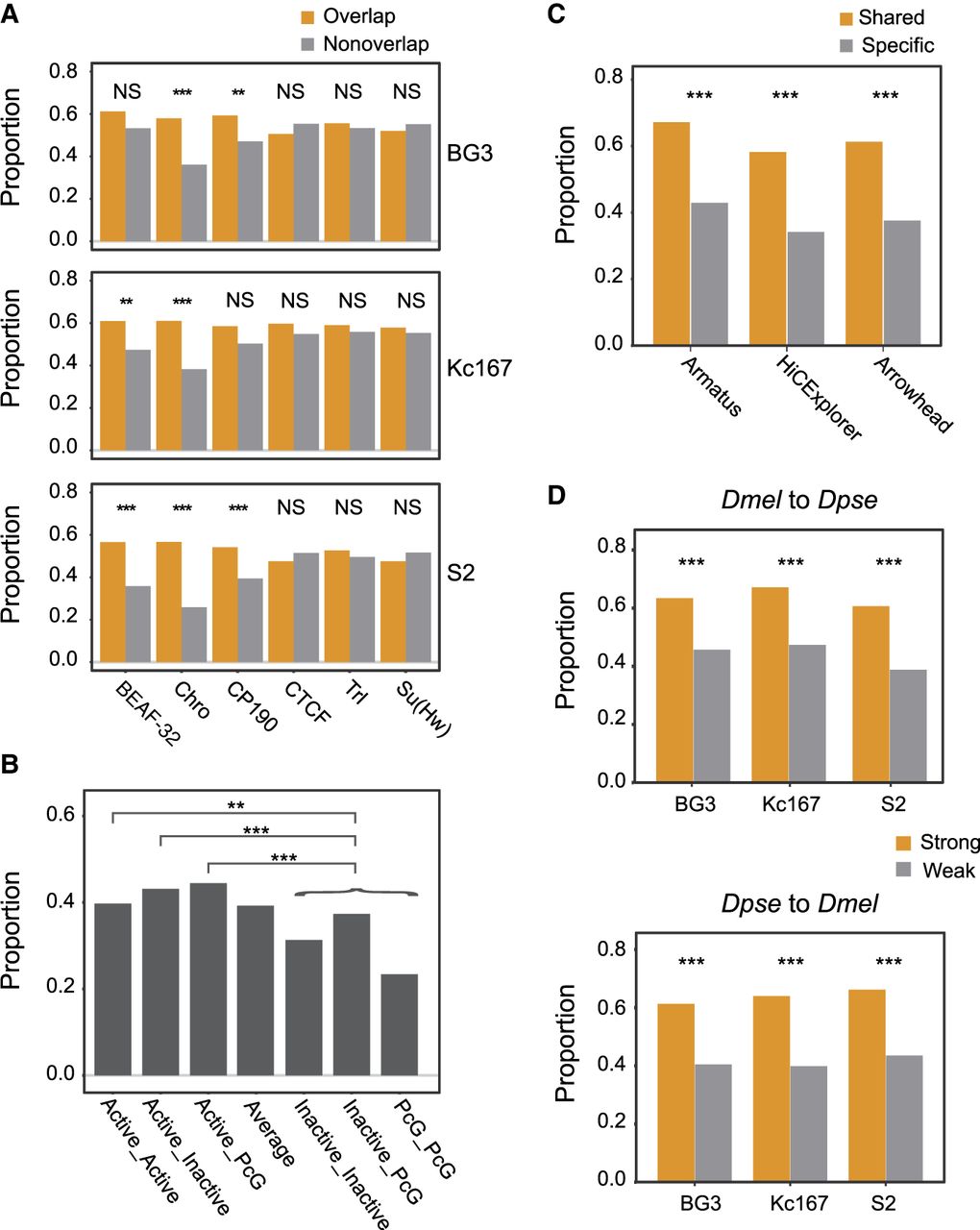
We also investigated the degree of conservation of the boundaries of TADs associated with different chromatin marks. By using the data from Ramírez et al. (2018), we found that boundaries associated with active TADs are more conserved than those of TADs enriched for inactive and PcG marks or those lacking chromatin marks altogether (Fig. 3B), which is consistent with the fact that active chromatin may play crucial roles in TAD formation (Ulianov et al. 2016).
Next, we partitioned TAD boundaries into cell-specific boundaries and boundaries observed in more than one cell line. HiCExplorer identified 921 D. melanogaster TAD boundaries shared by at least two cell lines, whereas 735 were cell specific (Supplemental Table S14). Of the 672/921 boundaries that we successfully lifted over to D. pseudoobscura, 58% (391/672) are conserved with D. pseudoobscura, which is significantly higher than cell-specific boundaries (P < 0.001, chi-square test), in which only 34% (165/483) are conserved with D. pseudoobscura (Fig. 3C). Similar results were also obtained for Arrowhead and Armatus calls (Fig. 3C). However, this pattern can also be caused by the potential underrepresentation of cell-specific boundaries in the D. pseudoobscura whole-body TAD set.
Finally, we compared stronger and weaker boundaries, wherein boundary strengths were categorized based on HiCExplorer TAD-separation score. Both types of boundaries tend to be observed in multiple cell lines, but weaker boundaries overlap more cell-specific boundaries (Supplemental Table S15). Hence, as indicated above, we expect stronger boundaries to be more conserved than weaker ones. We indeed observed this in the comparisons between D. pseudoobscura and each of the three D. melanogaster cell lines, as well as in the reciprocal comparisons (Fig. 3D).
The potential roles of TADs in Drosophila gene regulation
To investigate the potential link between TADs and gene regulation, we compared expression profiles across seven tissues from both sexes for 10,921 one-to-one orthologs between D. melanogaster and D. pseudoobscura (Yang et al. 2018). The orthologs were classified by their locations with respect to TADs or TAD boundaries (Juicer call), covering the following categories: (1) genes inside versus outside TADs, (2) genes inside conserved versus nonconserved TADs, (3) genes inside cell-specific versus those shared by multiple cell types TADs, (4) genes within 10 kbp of conserved versus nonconserved TAD boundaries, and (5) genes within 10 kbp of cell-specific TAD boundaries and those shared by multiple-cell-type TAD boundaries. To measure expression divergence, we calculated both the Euclidean distance and Pearson's correlation coefficient distance (Pereira et al. 2009). Both distances show that comparisons made using partitions from 1 through 3 above were statistically significant, whereas those made using 4 and 5 above (those involving proximity to TAD boundaries) were not (Fig. 4A; Supplemental Fig. S9A). These results show that TADs correlate with evolutionary stability of gene expression because genes inside TADs, conserved TADs, and TADs that are shared by multiple cell types all show significantly lower expression divergence than their corresponding counterparts, respectively.
The potential roles of TADs in gene regulation in Drosophila. (A) Expression divergence measured by Euclidean distance for one-to-one orthologs between D. melanogaster and D. pseudoobscura. (B) Expression variation measured by Euclidean distance for the same gene sets used in the above interspecific comparison between two D. melanogaster strains, OreR and w1118. (C) Physical overlap of long genes and TADs in D. melanogaster and D. pseudoobscura. (D) Representative GO biological process terms significantly enriched among the 127 long genes that constitute their own TADs in D. melanogaster. (E) Expression profile of the 127 long genes across eight tissues in both male and female in D. melanogaster. (AC) Abdomen without digestive or reproductive system; (DG) digestive plus excretory system; (GE) genitalia; (GO) gonad; (HD) head; (RE) reproductive system without gonad; (TX) thorax without digestive system; (WB) whole body; (F) female; (M) male.

We also considered the possibility that the stability of gene expression in annotated or conserved TAD regions is driven by gene content rather than TADs per se. To test this, we repeated the above analyses using the same gene set (10,921) between two D. melanogaster strains, OreR and w1118, with expression data across eight tissues from both sexes (Yang et al. 2018). This intraspecies analysis (assuming intraspecific TAD differences can be largely neglected) revealed similar patterns as those of cross-species comparisons (Fig. 4B; Supplemental Fig. S9B), suggesting that genes constrained in the annotated or conserved TAD regions indeed tend to be more stable.
We further investigated the link between the gene structures and TADs by focusing on protein-coding genes >50 kbp. Of 311 of those long genes (>50 kbp) in D. melanogaster, which span 30.35 Mb of the genome, we found that TAD boundaries are significantly depleted inside genes compared with the genome-wide background (143/1113 S2 boundaries; P = 4.86 × 10−15, proportion test against 30.35/134 Mb, or 0.226). Only 17 long genes contained TAD boundaries inside them in all three cell types (Fig. 4C). One hundred twenty-nine long genes (Supplemental Table S16), comprising 15 Mb of the D. melanogaster genome, individually occupied an intact TAD predicted in at least one of the three cell lines or tools (Supplemental Fig. S10). In D. pseudoobscura, we identified 338 long genes (>50 kbp), but we found relatively fewer long genes (73) that spanned full TADs (Supplemental Table S17). This may be because D. pseudoobscura TADs were annotated only in the whole body, whereas TADs in D. melanogaster come from three cell lines. This observation raises the possibility that some TADs emerge only in certain situations (e.g., cell types or developmental stages) to regulate their corresponding genes. This prediction is supported by Gene Ontology analysis (Supplemental Methods), which shows that functions of the long genes are enriched for development processes, particularly those affecting the nervous system (Fig. 4D), consistent with the observation that they are preferentially expressed in head (Fig. 4E; Supplemental Fig. S11).
Evolutionary genome rearrangement breakpoints coincide with Drosophila TAD boundaries
Conservation of a considerable fraction of TADs between two distantly related Drosophila species prompted us to investigate whether TADs are evolutionarily constrained. To this end, we identified 1061 genome rearrangement breakpoints between D. melanogaster and D. pseudoobscura (Fig. 5A; Supplemental Fig. S12) and found that they are enriched at TAD boundaries (257/1061; P < 2.2 × 10−16, proportion test against 11.13 Mb S2 HiCExplorer boundary regions as a proportion of total 134 Mb genome size, or 0.083), consistent with previous results (Lazar et al. 2018; Fishman et al. 2019).
Evolutionary genome rearrangement breakpoints are enriched at Drosophila TAD boundaries. (A) Synteny map between D. melanogaster 2L and D. pseudoobscura Chr 4. Tracks are as follows: (a) HiCExperor TAD boundaries annotated at restriction fragment resolution, (b) 10-kbp resolution, and (c) synteny breakpoints. (B) Phylogeny of 17 Drosophila species. Estimated divergence times are obtained from Thomas and Hahn (2017) except for Drosophila triauraria. (C) Distribution of genome rearrangement breakpoints between D. melanogaster and four other Drosophila species along TAD regions. (D) Distribution of genome rearrangement breakpoints between D. pseudoobscura and four species along TAD regions. (E) Conservation of TADs in an inverted genomic segment (D. melanogaster 2L: 8.55–8.95 Mb) between D. melanogaster and D. pseudoobscura.

We extended the analysis to 17 Drosophila species, spanning 72 million years of evolution (Fig. 5B; Thomas and Hahn 2017). All these species possess highly contiguous genome assemblies with contig N50 >4 Mb (Mahajan et al. 2018; Miller et al. 2018), permitting reliable identification of genome rearrangement breakpoints. Across comparisons between D. melanogaster and the other 16 species, we identified from 108 to 1180 synteny and from 10 to 314 inversion breakpoints (Supplemental Table S18). For most comparisons, we observed these breakpoints were enriched at TAD boundaries, whereas the frequency of breakpoints was depleted within TADs (Fig. 5C; Supplemental Fig. S13A). However, in comparisons between D. melanogaster and its closest relatives (D. sechellia, D. mauritiana, and D. simulans), the small number of events likely did not offer enough power to observe this pattern.
We repeated this analysis using D. pseudoobscura as the reference, identifying from 259 to 1242 synteny and from 60 to 359 inversion breakpoints in the 16 analogous comparisons (Supplemental Table S18). We observed the same pattern as above (Fig. 5D; Supplemental Fig. S13B). Such enrichment of rearrangement breakpoints at TAD boundaries suggests that a fraction of TADs are evolutionarily constrained even among genomes as extensively rearranged as those in the genus Drosophila. For example, three TADs are well preserved on a ∼450-kb inverted genomic segment between D. melanogaster and D. pseudoobscura across 49 million years of evolution (Fig. 5E).
TADs shape structural genomic variants at their boundaries in Drosophila
To investigate the potential role of TADs in shaping patterns of structural genomic variants, we obtained two comprehensive SV data sets (comprising deletions, TE insertions, non-TE insertions, and tandem duplications [TDs]), both based on reference-quality genome assemblies (Fig. 6A), which include (1) a polymorphic SV data set (Fig. 6B) from 14 D. melanogaster strains (Chakraborty et al. 2019) and (2) a divergence SV data set spanning ∼3 million years of evolution between D. melanogaster and the three members of the D. simulans species complex (Fig. 6C; Chakraborty et al. 2020). Deletions and insertions were polarized using D. erecta and D. yakuba as outgroups. The resulting unfolded allele frequency spectrum (Fig. 6D) shows that, in the polymorphic data set, most SVs are observed in only a single strain, with TE insertions showing the greatest proportion (∼92%) of rare variants than other types. However, a considerable proportion (35%–63%) of interspecific SVs are present in at least two species (Fig. 6E), suggesting that these SVs occurred before at least one species-splitting event in the D. simulans clade and therefore have existed for a relatively longer period of time than intraspecies SVs.
Patterns of structural variants at Drosophila TAD boundaries. (A) Highly contiguous genome assemblies from 14 D. melanogaster strains and three D. simulans clade species, together with two outgroup species, D. erecta and D. yakuba. (B) Nonredundant SVs, including TE insertions, tandem duplications (DUP), non-TE insertions (INS; “S” represents insertions size range from 1-10 bp and “L” represents insertions size range from 11 bp to 20 kbp), and deletions (DEL; “S” for 1–10 bp and “L” for 11 bp to 2 kbp) identified from the 14 D. melanogaster strains. (C) Nonredundant SVs identified in the three D. simulans clade species. (D) The unfolded site frequency spectrum of SVs from 14 D. melanogaster strains. (E) Phylogenetic profiling of SVs among the three D. simulans clade species. (F) Tests of purifying selection on SVs at TAD boundaries using Fudenberg and Pollard's method. (G) Odds ratios of 2 × 2 contingency tables with margins categorizing polymorphism/divergence and boundary/nonboundary mutations. Confidence intervals are calculated from the Fisher's exact test results. (H) Deletions from both data sets are depleted at the TAD boundaries. (I) Non-TE insertions from both data sets are depleted at the TAD boundaries. Red and black lines represent larger and shorter variants, respectively. (***) P < 1 × 10−4, permutation test (Supplemental Fig. S15).

By using these two data sets, we sought to assess the mode of selection on SVs at Drosophila TAD boundaries by exploiting the method described previously (Fudenberg and Pollard 2019). We used a conservative set of 2156 TAD boundaries in the euchromatin regions (Supplemental Table S19) that are shared between two independent studies (Ramírez et al. 2018; Wang et al. 2018). We find that both deletions and small non-TE insertions are depleted at TAD boundaries in both polymorphic and divergence SV data sets (Fig. 6F; Supplemental Fig. S14), consistent with that purifying selection may act to remove them to prevent disruption of TAD boundaries. A contrast was observed for TDs. TDs from 14 D. melanogaster strains are depleted at TAD boundaries compared with genome-wide background (181/3606 breakpoints; P = 2.83 × 10−8, proportion test against 8.6-Mb boundary regions as a proportion of total 117-Mb euchromatin regions, or 0,074), whereas TDs from the three D. simulans clade species are significantly enriched at TAD boundaries (287/2582 breakpoints; P = 3.63 × 10−13, proportion test against 0.074). The enrichment of TDs at TAD boundaries are also observed in the comparison between D. pseudoobscura and D. miranda (293/2728 breakpoints, P < 2.2 × 10−16, permutation test against 8.86 Mb/133.62 Mb) (Fig. 6F; Supplemental Table S20). This excess divergence relative to polymorphism may indicate the action of adaptive selection (Andolfatto 2005). To quantify this excess for all other genomic variants studied, we calculated the odds ratios by dividing the ratio of genomic variants occurring at TAD boundary regions versus the rest of euchromatin regions in the divergence data set by the same ratio computed from polymorphism (Fig. 6G). The highest excess value of TDs (287/2295 in divergence TDs vs. 181/3425 in polymorphic TDs; P < 2.2 × 10−16, Fisher's exact test) suggests that TDs at TAD boundaries might have evolved under adaptive natural selection.
Enrichment of TDs at TAD boundaries is consistent with previous observations that TAD boundaries tend to act as relatively frequent targets of duplications (Sadowski et al. 2019) and coduplicate with super enhancers (Gong et al. 2018). To determine what kinds of sequences are associated with duplicated TAD boundaries, we inspected 181 boundary TDs in the three D. simulans clade species, finding that they rarely overlap large-scale genome rearrangement breakpoints, instead ∼88% (160/181) of them overlap genes. These genes are weakly enriched for constitutive genes (i.e., genes that are expressed in most or all cells of an organism; 70/169; P = 0.018, proportion test against the expected proportion of 5854/17,473) (Supplemental Methods), consistent with the previous observation that TAD boundaries are enriched in housekeeping genes (Hug et al. 2017).
Finally, with the large number of SVs we identified in our data sets, we also investigated SVs in the surrounding TAD boundary regions. Confirming the observations above, deletions and non-TE insertions are broadly depleted around TAD boundaries with peaks at the boundaries (Fig. 6H,I). Larger deletions in the polymorphic data set are more depleted at TAD boundaries, implying larger deletions may be more deleterious to TAD boundaries (Fig. 6H). It is worth noting that TE insertions show complex patterns at TAD boundaries. For example, long terminal repeat retrotransposons (LTRs) and long interspersed nuclear elements (LINEs) are strongly depleted at TAD boundaries (P < 1.0 × 10−4, permutation test) in the polymorphic data set, but such a pattern is not observed in the divergence data set or for DNA-type TEs (Supplemental Fig. S15).
Discussion
Our knowledge of 3D genome evolution remains limited (Yang et al. 2019). To interrogate the evolutionary patterns of genome topology and its potential association with genome structure and function, we generated a reference-quality genome assembly and high-resolution Hi-C data for D. pseudoobscura.
Although our D. pseudoobscura Hi-C data were obtained from whole-body samples, we observed high consistency with several biological features known to be associated with TADs, regardless of the specific approaches used in their annotation. For example, our TAD annotations are correlated with epigenetic states (e.g., H3K4me3 and H3K27me3), and their boundaries are enriched for insulator binding sites (e.g., CTCF and BEAF-32) and open and active chromatin marks (Fig. 1). These observations are consistent with the fact that TADs are a largely invariant feature across tissues in a given organism (Ulianov et al. 2016). Although CTCF appears not to be a major TAD boundary definition protein in Drosophila (Szabo et al. 2019), we nevertheless observed enrichment of CTCF at our TAD boundaries, likely because it connects TAD borders in a cell-specific manner in Drosophila (Chathoth and Zabet 2019).
Our analysis revealed that TADs are conserved across at least 30-40 percent of the genomes between D. melanogaster and D. pseudoobscura. This rate is comparable to that observed between the much more closely related comparison between humans and their closest sister species, chimpanzees (∼43%) (Eres et al. 2019). The conservation we observe is substantially higher than that among three distantly related Drosophila species: D. melanogaster, Drosophila busckii, and Drosophila virilis (∼10%) (Renschler et al. 2019). Such incongruity is perhaps explained by differences in the quality of genome assemblies, depth of Hi-C data, or the evolutionary distance between species comparisons. Despite the difference, both results showed that a substantial proportion of TADs persists for long periods of time during evolution, suggesting they are functionally relevant. It is worth noting that our study likely still underestimates conservation. Our estimates were derived from pairwise comparisons between three distinct cell lines (Kc167, S2, and BG3) in D. melanogaster but whole body in D. pseudoobscura. Given extensive cell- and allele-specific variability of TADs observed using single-cell Hi-C (Nagano et al. 2013) and super-resolution fluorescence in situ hybridization (FISH) imaging approaches (Bintu et al. 2018), TADs identified in whole-body samples might represent topologies averaged across multiple tissues and millions of cells. Thus, many cell-specific, tissue-specific, or developmental stage–specific TADs may be underrepresented. Future experiments that carefully match samples (e.g., the same cell types or tissues) may provide a path to address this problem.
The role of TADs in gene regulation remains a matter of active research. Recent thinking suggests a reciprocal interplay between spatial genome organization and transcription, in which each is able to modulate or reinforce the activity of the other (van Steensel and Furlong 2019). Our results and others (Krefting et al. 2018) have revealed that the evolutionary stability of TADs correlates to constraint on gene expression, suggesting TADs may play roles in gene regulation. This effect is potentially confounded by the properties of the gene content in conserved TADs, although these two possibilities are by no means mutually exclusive. For example, the pattern of constraint we see in gene regulation is already established on intraspecific timescales in D. melanogaster, potentially before enough time has elapsed to establish variation between constraint in TADs.
Our finding that a large proportion of long genes coincides with entire TADs implies that transcription may be one of the deterministic factors for the establishment and maintenance of spatial genomic organization or, conversely, that TADs are important in the regulation of long genes. Such gene-level chromatin domains are reminiscent of self-loop structures of genes found in Arabidopsis thaliana (Liu et al. 2016) and gene crumples in Saccharomyces cerevisiae (Hsieh et al. 2015). Moreover, such gene-level domains are more likely to be cell-, tissue-, or developmental stage-specific because we detected substantially more (129 or 46%) genes in individual cell lines in D. melanogaster than in whole-body samples (73 or 26%) in D. pseudoobscura.
Our analyses, in combination with previous works (Krefting et al. 2018; Lazar et al. 2018; Fishman et al. 2019; Renschler et al. 2019), show that genome rearrangement breakpoints acquired during evolution preferentially occur at TAD borders, suggesting that rearrangements resulting in disruption of TAD integrity are subjected to negative selection. Moreover, it suggests that TADs tend to evolve as intact structural units in genome shuffling, probably owing to their putative functional constraint. Nevertheless, the nonrandom distribution of chromosomal breaks can also be explained by the “fragile regions” model that breaks of chromosome occur at a higher frequency at TAD borders than genome background (Berthelot et al. 2015). Further experiments designed to characterize recurrent breakpoints across the genome would be necessary to clarify the above hypotheses.
We also found evidence for selection acting on structural genomic variants at TAD boundaries. Recent studies have found that deletions are depleted at TAD boundaries in human populations (Sadowski et al. 2019) and in humans’ close relatives, apes (Fudenberg and Pollard 2019), as well as in cancer genomes (Akdemir et al. 2020). In Drosophila, we observed the same pattern for deletions and non-TE insertions, suggesting that these common SVs are subject to purifying selection. Unlike the above two types of SVs, patterns of TE insertions not only are different in their classes but also differ in evolutionary timescales. In polymorphic SV data sets, LTRs and LINEs are strongly depleted at TAD boundaries, whereas DNA-type TE is slightly enriched at TAD boundaries (Supplemental Table S15), suggesting they are under different selective pressure. Furthermore, such patterns were not observed in the divergence SV data set, possibly because most of the older deleterious TE insertions have already been eliminated uniformly across the genomes of the D. simulans complex species. TAD boundaries seem to appear largely in gene-dense, chromatin-accessible, and transcribed regions where enriched in active chromatin marks (Szabo et al. 2019). For example, ∼77% of TAD boundaries annotated in D. melanogaster overlap promoters (Ramírez et al. 2018). Thus, it remains unclear which functional aspects are principal factors governing constraints of SVs at TAD boundaries.
The finding that divergence of TDs is elevated at TAD boundaries relative to elsewhere when both are normalized by levels of polymorphism (Fig. 6G) suggests that tandem duplicates at TAD boundaries are fixed at higher rates, although this imbalance in the odds ratio could also stem from a deviation in any of the four terms. The absence of enrichment of TDs at TAD boundaries in the polymorphism data suggests that this finding is unlikely to be mutationally driven. Therefore, we propose that adaptation could be driving up the divergence of tandem duplicates at TAD boundaries. One intuitive reason for this suggestion may be that duplicated boundary sequences, such as insulator binding sites, may strengthen topological domain borders, thereby reinforcing the stability of chromatin domains. This is consistent with the hypothesis that duplications may be an important evolutionary mechanism of spatial genome organization (Sadowski et al. 2019). Similarly, the fact that TAD boundary duplications largely overlap with functional regulatory elements and genes argues for further examining the forces shaping enrichment of TDs at TAD boundaries. Collectively, our findings offer novel insight into the evolutionary significance of spatial genome organization in shaping patterns of large-scale chromosomal rearrangements, common SVs, and gene expression.
Methods
Fly strain and genome sequencing
The sequenced D. pseudoobscura strain (MV-25-SWS-2005) was initially collected at Mesa Verde, Colorado (latitude 37d 18′ 0′′ N, longitude 108d 24′ 58′′ W) in July 2005 by Stephen W. Schaeffer. The strain was subsequently inbred. DNA was extracted from adult females following a previously published protocol (Chakraborty et al. 2016). DNA was sheared using 21-gauge needles and size-selected using the 30- to 80-kbp cutoff on Blue Pippin (Sage Science). Size-selected DNA was sequenced on 10 SMRT cells using the PacBio sequel platform. Illumina paired-end (2 × 150 bp) reads were generated on HiSeq 4000 using the same DNA that was used for PacBio sequencing. PacBio long reads were assembled with Canu v1.7 (Koren et al. 2017). After the removal of redundant contigs and gap filling using raw reads with finisherSC (Lam et al. 2015), the assembly was polished twice with Arrow (Smrtanalysis 5.1.0) and three times with Pilon (Walker et al. 2014). TEs were annotated using the EDTA pipeline (Ou et al. 2019). Gene models were annotated using MAKER (version 2.31.8) (Campbell et al. 2014). More details described in the Supplemental Methods.
Hi-C experiments
Hi-C experiments were performed by Arima Genomics (https://arimagenomics.com/) with adult female flies according to the Arima-HiC protocol described in the Arima-HiC kit (P/N: A510008) with minor modifications to the crosslinking protocol (for details, see Supplemental Methods).
Hi-C data processing and TAD annotation
Juicer (Durand et al. 2016) and HiCExplorer (Ramírez et al. 2018) were used to process Hi-C data from raw reads to interaction maps. Arrowhead from Juicer package, Armatus (Filippova et al. 2014), and HiCExplorer were used to annotate TADs, each with different combinations of parameters. The output was compared and inspected visually based on chromatin interaction maps (Supplemental Fig. S16) using HiCPlotter (Akdemir and Chin 2015) to determine the optimal parameters (Supplemental Table S21).
ChIP-seq and ATAC-seq data analysis
All short-read alignments were performed against our D. pseudoobscura genome using Bowtie 2 v2.2.7 (Langmead and Salzberg 2012). ChIP-seq peak calling was performed using MACS2 (version 2.0.10) (Zhang et al. 2008) with the default parameters. ChIP-seq normalization was performed using bamCompare from the deepTools suite (version 3.2.1) (Ramírez et al. 2016) with the following setting: “‐‐binSize 10 ‐‐operation log2 ‐‐minMappingQuality 30 ‐‐skipNonCoveredRegions ‐‐ignoreDuplicates”. Read coverage of ATAC-seq was computed using deepTools bamCoverage for a bin size of 10 bp. To generate metaregion plots (Fig. 1D–F) of ChIP-seq/ATAC-seq signals or frequency of insulator binding sites surrounding TAD boundaries, a matrix Aij was generated for each data set using deepTools computeMatrix and Perl scripts, in which each row represents a boundary and each column (j ∈[−40,40]) represents the signal value in a 1-kbp nonoverlapping bin within 40 kbp of the downstream and upstream flanking regions of that boundary. For CTCF and BEAF-32 binding sites, we summed values from columns of Aij into a vector in which each element represents the signal value for the corresponding 1-kbp bin. For ChIP-seq and ATAC-seq data, we averaged values from columns of Aij into a vector. To assess the significance of each signal at TAD boundaries, we generated 10,000 random samples of simulated TAD boundaries with the number and chromosome distribution confined by the observed data set using BEDTools shuffle (version 2.25.0) (Quinlan and Hall 2010). We then computed the sampling distribution of each signal value around TAD boundaries in the same way as described above for actual boundaries and determined the P-values.
The preprocessed ChIP data for D. melanogaster were obtained from the modENCODE Consortium (The modENCODE Consortium et al. 2010; http://www.modencode.org/).
Identification of conserved TAD features and significance tests
To identify conserved TAD features (i.e., body and boundary), genomic coordinates were converted between species using the UCSC liftOver tool and the Dmel-Dpse chain file generated in this study. To be successfully lifted over, features in one species require a 25% minimum ratio of bases (−minMatch = 0.25) for body and one-third for boundary (−minMatch = 0.33) to be remapped in the other species, and the size difference should not exceed 50% for body and 100% for boundary. Conserved TAD bodies were determined using BEDTools intersect with the parameters: -F 0.8 -f 0.8, which requires at least 80% reciprocal overlap in the corresponding intervals in both species. For boundaries, we considered any overlap as indicative of conservation.
To determine if the observed conservation of TAD features is statistically significant, we tested two null hypotheses. First, we assumed that the locations of TAD features across the genome are completely independent between separate species. To test this, we simulated 10,000 random samples of TAD features in one species and computed the sampling distribution of conservation with the other species. The P-values were then determined by the permutation distributions or Fisher's exact tests based on the observed and expected (mean of the 10,000 simulations) number of lifted and conserved TAD features. As an alternative null hypothesis for the TAD body, we assumed that TADs are completely conserved across the genome between species only when chromosomal rearrangements can disrupt them. To test this, we simulated 10,000 sets of genome shuffling by random fragmentation in each species. The size distribution of each sample of genome fragments requires matching the actual synteny blocks between D. melanogaster and D. pseudoobscura. A TAD from the first species was considered to be conserved if it is successfully lifted and at least 80% of the converted genomic coordinate overlaps with any of the simulated genome fragments in the second species. Then, the sampling distribution of the conservation was used to determine the P-values.
Gene expression data analysis
The preprocessed expression data were obtained from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo) under accession ID GSE99574. Orthologs were obtained from FlyBase (https://flybase.org/) ortholog gene sets. After filtering, 10,921 of the 13,638 Dmel-Dpse orthologs we retrieved are in a one-to-one relationship and have expression data. To measure expression divergence, we computed both Euclidean distance and Pearson's correlation coefficient distance following the formulas previously described (Pereira et al. 2009).
Assembly-based SV detection
SV calling was performed following our custom pipeline (Liao et al. 2018; Kou et al. 2020) based on the LASTZ/CHAIN/NET workflow (for a more detailed description of the pipeline, see Supplemental Methods; Schwartz et al. 2003; Harris 2007). The pipeline is available on GitHub (https://github.com/yiliao1022/LASTZ_SV_pipeline).
Identification and analysis of evolutionary chromosomal rearrangement breakpoints
Pairwise genome alignments were performed against D. melanogaster and D. pseudoobscura genome, respectively, using LASTZ (version 1.04). The resulting alignments were then processed with axtChain/chainNet/netSyntenic tools to get the netSyntenic files, which were used as input in our custom Perl script Synbreaks.pl to identify chromosomal rearrangement breakpoints. Breakpoints were classified into two categories: (1) synteny breaks if they were obtained from “top,” “syn,” or “NonSyn” fills and (2) inversion breaks if they were obtained from “inv” fills. We excluded breaks that were identified from synteny blocks of a size <10 kbp and near the terminal regions (<10 kbp) of long contigs in our analysis, because they are more likely introduced by assembling artifacts.
To quantify the distribution of rearrangement breakpoints along the TADs, we followed a previously described method (Krefting et al. 2018). Briefly, each TAD domain was extended by 50% of its size on each side, and the resulting interval was subdivided into 20 equal-sized bins. The occurrence of breakpoints was then summed over bins for all TADs to generate a vector in which each element represents one of the 20 bins. Additionally, we generated 100 sets of random breakpoints as background control.
Selection of SVs at TAD boundaries
We measured the relative abundance of SVs at TAD boundaries following a previously described method (Supplemental Methods; Fudenberg and Pollard 2019). We also permuted 10,000 sets of TAD boundaries across the genome, excluding heterochromatic regions, to generate the background distribution of the relative abundance of SVs at TAD boundaries for statistical tests.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA596268. The code that reproduces analyses from the manuscript is available as Supplemental Code and at GitHub (https://github.com/yiliao1022/TADEvoDrosophila).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was funded by the National Institutes of Health (R01GM123303), National Science Foundation (IOS-1656260), and start-up funding from the University of California, Irvine to J.J.E. We thank the Genomics High Throughput Facility at University of California, Irvine (UCI) and Arima Genomics San Diego for expert service. We also thank Luna Thanh Ngo for her help with data collection and management. We thank Stephen W. Schaeffer for supplying the strain sequenced in the study.
Author contributions: Y.L. and J.J.E. conceived the study and designed the research. M.C. contributed to the genomic data collection. M.C. and Y.L. generated the genome assembly. Y.L., X.Z., and J.J.E. analyzed the data and interpreted the results. Y.L. wrote the original draft. All authors reviewed and edited the manuscript.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.266130.120.
References
- ↵Akdemir KC, Chin L. 2015. HiCPlotter integrates genomic data with interaction matrices. Genome Biol 16: 198. 10.1186/s13059-015-0767-1
- ↵Akdemir KC, Le VT, Chandran S, Li Y, Verhaak RG, Beroukhim R, Campbell PJ, Chin L, Dixon JR, Futreal PA, 2020. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat Genet 52: 294–305. 10.1038/s41588-019-0564-y
- ↵Andolfatto P. 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437: 1149–1152. 10.1038/nature04107
- ↵Arnold CD, Gerlach D, Spies D, Matts JA, Sytnikova YA, Pagani M, Lau NC, Stark A. 2014. Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat Genet 46: 685–692. 10.1038/ng.3009
- ↵Bartolomé C, Charlesworth B. 2006. Rates and patterns of chromosomal evolution in Drosophila pseudoobscura and D. miranda. Genetics 173: 779–791. 10.1534/genetics.105.054585
- ↵Berthelot C, Muffato M, Abecassis J, Roest Crollius H. 2015. The 3D organization of chromatin explains evolutionary fragile genomic regions. Cell Rep 10: 1913–1924. 10.1016/j.celrep.2015.02.046
- ↵Bintu B, Mateo LJ, Su J-H, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang X. 2018. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362: eaau1783. 10.1126/science.aau1783
- ↵Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J-P, Tanay A, 2017. Multiscale 3D genome rewiring during mouse neural development. Cell 171: 557–572.e24. 10.1016/j.cell.2017.09.043
- ↵Bracewell R, Chatla K, Nalley MJ, Bachtrog D. 2019. Dynamic turnover of centromeres drives karyotype evolution in Drosophila. eLife 8: e49002. 10.7554/eLife.49002
- ↵Campbell MS, Holt C, Moore B, Yandell M. 2014. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinformatics 48: 188. 10.1002/0471250953.bi0411s48
- ↵Chakraborty M, Baldwin-Brown JG, Long AD, Emerson JJ. 2016. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res 44: e147. 10.1093/nar/gkw654
- ↵Chakraborty M, Emerson JJ, Macdonald SJ, Long AD. 2019. Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits. Nat Commun 10: 4872. 10.1038/s41467-019-12884-1
- ↵Chakraborty M, Chang C-H, Khost DE, Vedanayagam J, Adrion JR, Liao Y, Montooth K, Meiklejohn CD, Larracuente AM, Emerson JJ. 2020. Evolution of genome structure in the Drosophila simulans species complex. Genome Res (this issue). 10.1101/gr.263442.120
- ↵Chathoth KT, Zabet NR. 2019. Chromatin architecture reorganization during neuronal cell differentiation in Drosophila genome. Genome Res 29: 613–625. 10.1101/gr.246710.118
- ↵Despang A, Schöpflin R, Franke M, Ali S, Jerković I, Paliou C, Chan W-L, Timmermann B, Wittler L, Vingron M, 2019. Functional dissection of the Sox9–Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nat Genet 51: 1263–1271. 10.1038/s41588-019-0466-z
- ↵Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. 10.1038/nature11082
- ↵Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, Aiden EL. 2016. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3: 95–98. 10.1016/j.cels.2016.07.002
- ↵English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, 2012. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One 7: e47768. 10.1371/journal.pone.0047768
- ↵Eres IE, Luo K, Hsiao CJ, Blake LE, Gilad Y. 2019. Reorganization of 3D genome structure may contribute to gene regulatory evolution in primates. PLoS Genet 15: e1008278. 10.1371/journal.pgen.1008278
- ↵Filippova D, Patro R, Duggal G, Kingsford C. 2014. Identification of alternative topological domains in chromatin. Algorithms Mol Biol 9: 14. 10.1186/1748-7188-9-14
- ↵Fishman V, Battulin N, Nuriddinov M, Maslova A, Zlotina A, Strunov A, Chervyakova D, Korablev A, Serov O, Krasikova A. 2019. 3D organization of chicken genome demonstrates evolutionary conservation of topologically associated domains and highlights unique architecture of erythrocytes’ chromatin. Nucleic Acids Res 47: 648–665. 10.1093/nar/gky1103
- ↵Forcato M, Nicoletti C, Pal K, Livi CM, Ferrari F, Bicciato S. 2017. Comparison of computational methods for Hi-C data analysis. Nat Methods 14: 679–685. 10.1038/nmeth.4325
- ↵Fudenberg G, Pollard KS. 2019. Chromatin features constrain structural variation across evolutionary timescales. Proc Natl Acad Sci 116: 2175–2180. 10.1073/pnas.1808631116
- ↵Ghavi-Helm Y, Jankowski A, Meiers S, Viales RR, Korbel JO, Furlong EEM. 2019. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat Genet 51: 1272–1282. 10.1038/s41588-019-0462-3
- ↵Gong Y, Lazaris C, Sakellaropoulos T, Lozano A, Kambadur P, Ntziachristos P, Aifantis I, Tsirigos A. 2018. Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries. Nat Commun 9: 542. 10.1038/s41467-018-03017-1
- ↵Harris RS. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, Pennsylvania State University, University Park, PA. https://etda.libraries.psu.edu/catalog/7971.
- ↵Hsieh T-HS, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. 2015. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 162: 108–119. 10.1016/j.cell.2015.05.048
- ↵Hug CB, Grimaldi AG, Kruse K, Vaquerizas JM. 2017. Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169: 216–228.e19. 10.1016/j.cell.2017.03.024
- ↵Huynh L, Hormozdiari F. 2019. TAD fusion score: discovery and ranking the contribution of deletions to genome structure. Genome Biol 20: 60. 10.1186/s13059-019-1666-7
- ↵Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27: 722–736. 10.1101/gr.215087.116
- ↵Kou Y, Liao Y, Toivainen T, Lv Y, Tian X, Emerson JJ, Gaut BS, Zhou Y. 2020. Evolutionary genomics of structural variation in Asian rice (Oryza sativa) domestication. Mol Biol Evol 37: 3507–3524. 10.1093/molbev/msaa185
- ↵Krefting J, Andrade-Navarro MA, Ibn-Salem J. 2018. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biol 16: 87. 10.1186/s12915-018-0556-x
- ↵Lam K-K, LaButti K, Khalak A, Tse D. 2015. FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics 31: 3207–3209. 10.1093/bioinformatics/btv280
- ↵Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923
- ↵Lazar NH, Nevonen KA, O'Connell B, McCann C, O'Neill RJ, Green RE, Meyer TJ, Okhovat M, Carbone L. 2018. Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res 28: 983–997. 10.1101/gr.233874.117
- ↵Le TBK, Imakaev MV, Mirny LA, Laub MT. 2013. High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342: 731–734. 10.1126/science.1242059
- ↵Li LL, Lyu X, Hou C, Takenaka N, Nguyen HQ, Ong C-T, Cubeñas-Potts C, Hu M, Lei EP, 2015. Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol Cell 58: 216–231. 10.1016/j.molcel.2015.02.023
- ↵Liao Y, Zhang X, Li B, Liu T, Chen J, Bai Z, Wang M, Shi J, Walling JG, Wing RA, 2018. Comparison of Oryza sativa and Oryza brachyantha genomes reveals selection-driven gene escape from the centromeric regions. Plant Cell 30: 1729–1744. 10.1105/tpc.18.00163
- ↵Liu C, Wang C, Wang G, Becker C, Zaidem M, Weigel D. 2016. Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res 26: 1057–1068. 10.1101/gr.204032.116
- ↵Liu C, Cheng Y-J, Wang J-W, Weigel D. 2017. Prominent topologically associated domains differentiate global chromatin packing in rice from Arabidopsis. Nat Plants 3: 742–748. 10.1038/s41477-017-0005-9
- ↵Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, 2015. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161: 1012–1025. 10.1016/j.cell.2015.04.004
- ↵Mahajan S, Wei KH-C, Nalley MJ, Gibilisco L, Bachtrog D. 2018. De novo assembly of a young Drosophila Y chromosome using single-molecule sequencing and chromatin conformation capture. PLoS Biol 16: e2006348. 10.1371/journal.pbio.2006348
- ↵Malko DB. 2006. Evolution of exon–intron structure and alternative splicing in fruit flies and malarial mosquito genomes. Genome Res 16: 505–509. 10.1101/gr.4236606
- ↵Miller DE, Staber C, Zeitlinger J, Hawley RS. 2018. Highly contiguous genome assemblies of 15 Drosophila species generated using nanopore sequencing. G3 8: 3131–3141. 10.1534/g3.118.200160
- ↵Mizuguchi T, Fudenberg G, Mehta S, Belton J-M, Taneja N, Folco HD, FitzGerald P, Dekker J, Mirny L, Barrowman J, 2014. Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature 516: 432–435. 10.1038/nature13833
- ↵The modENCODE Consortium, Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, 2010. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330: 1787–1797. 10.1126/science.1198374
- ↵Muller HJ. 1940. Bearings of the ‘Drosophila’ work on systematics. In The New Systematics (ed. Huxley J), pp. 185–268. Clarendon Press, Oxford, UK.
- ↵Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. 2013. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502: 59–64. 10.1038/nature12593
- ↵Ni X, Zhang YE, Nègre N, Chen S, Long M, White KP. 2012. Adaptive evolution and the birth of CTCF binding sites in the Drosophila genome. PLoS Biol 10: e1001420. 10.1371/journal.pbio.1001420
- ↵Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. 10.1038/nature11049
- ↵Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ, Lugo CSB, Elliott TA, Ware D, Peterson T, 2019. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20: 275. 10.1186/s13059-019-1905-y
- ↵Pereira V, Waxman D, Eyre-Walker A. 2009. A problem with the correlation coefficient as a measure of gene expression divergence. Genetics 183: 1597–1600. 10.1534/genetics.109.110247
- ↵Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, 2014. Topologically associating domains are stable units of replication-timing regulation. Nature 515: 402–405. 10.1038/nature13986
- ↵Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033
- ↵Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. 10.1093/nar/gkw257
- ↵Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T. 2018. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9: 189. 10.1038/s41467-017-02525-w
- ↵Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680. 10.1016/j.cell.2014.11.021
- ↵Renschler G, Richard G, Valsecchi CIK, Toscano S, Arrigoni L, Ramírez F, Akhtar A. 2019. Hi-C guided assemblies reveal conserved regulatory topologies on X and autosomes despite extensive genome shuffling. Genes Dev 33: 1591–1612. 10.1101/gad.328971.119
- ↵Richards S, Liu Y, Bettencourt BR, Hradecky P, Letovsky S, Nielsen R, Thornton K, Hubisz MJ, Chen R, Meisel RP, 2005. Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res 15: 1–18. 10.1101/gr.3059305
- ↵Rowley MJ, Corces VG. 2018. Organizational principles of 3D genome architecture. Nat Rev Genet 19: 789–800. 10.1038/s41576-018-0060-8
- ↵Sadowski M, Kraft A, Szalaj P, Wlasnowolski M, Tang Z, Ruan Y, Plewczynski D. 2019. Spatial chromatin architecture alteration by structural variations in human genomes at the population scale. Genome Biol 20: 148. 10.1186/s13059-019-1728-x
- ↵Schaeffer SW. 2018. Muller “elements” in Drosophila: how the search for the genetic basis for speciation led to the birth of comparative genomics. Genetics 210: 3–13. 10.1534/genetics.118.301084
- ↵Schmitt AD, Hu M, Ren B. 2016. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews Molecular Cell Biology 17: 743–755. 10.1038/nrm.2016.104
- ↵Schoenfelder S, Fraser P. 2019. Long-range enhancer–promoter contacts in gene expression control. Nat Rev Genet 20: 437–455. 10.1038/s41576-019-0128-0
- ↵Schuettengruber B, Oded Elkayam N, Sexton T, Entrevan M, Stern S, Thomas A, Yaffe E, Parrinello H, Tanay A, Cavalli G. 2014. Cooperativity, specificity, and evolutionary stability of Polycomb targeting in Drosophila. Cell Rep 9: 219–233. 10.1016/j.celrep.2014.08.072
- ↵Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W. 2003. Human–mouse alignments with BLASTZ. Genome Res 13: 103–107. 10.1101/gr.809403
- ↵Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. 2012. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148: 458–472. 10.1016/j.cell.2012.01.010
- ↵Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. 10.1093/bioinformatics/btv351
- ↵Szabo Q, Jost D, Chang J-M, Cattoni DI, Papadopoulos GL, Bonev B, Sexton T, Gurgo J, Jacquier C, Nollmann M, 2018. TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci Adv 4: eaar8082. 10.1126/sciadv.aar8082
- ↵Szabo Q, Bantignies F, Cavalli G. 2019. Principles of genome folding into topologically associating domains. Sci Adv 5: eaaw1668. 10.1126/sciadv.aaw1668
- ↵Thomas G, Hahn M. 2017. Drosophila 25 species phylogeny. https://figshare.com/articles/Drosophila_25_species_phylogeny/5450602.
- ↵Ulianov SV, Khrameeva EE, Gavrilov AA, Flyamer IM, Kos P, Mikhaleva EA, Penin AA, Logacheva MD, Imakaev MV, Chertovich A, 2016. Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res 26: 70–84. 10.1101/gr.196006.115
- ↵van Steensel B, Furlong EEM. 2019. The role of transcription in shaping the spatial organization of the genome. Nat Rev Mol Cell Biol 20: 327–337. 10.1038/s41580-019-0114-6
- ↵Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. 10.1371/journal.pone.0112963
- ↵Wang Q, Sun Q, Czajkowsky DM, Shao Z. 2018. Sub-kb Hi-C in D. melanogaster reveals conserved characteristics of TADs between insect and mammalian cells. Nat Commun 9: 188. 10.1038/s41467-017-02526-9
- ↵Yang J, Ramos E, Corces VG. 2012. The BEAF-32 insulator coordinates genome organization and function during the evolution of Drosophila species. Genome Res 22: 2199–2207. 10.1101/gr.142125.112
- ↵Yang H, Jaime M, Polihronakis M, Kanegawa K, Markow T, Kaneshiro K, Oliver B. 2018. Re-annotation of eight Drosophila genomes. Life Sci Alliance 1: e201800156. 10.26508/lsa.201800156
- ↵Yang Y, Zhang Y, Ren B, Dixon JR, Ma J. 2019. Comparing 3D genome organization in multiple species using phylo-HMRF. Cell Syst 8: 494–505.e14. 10.1016/j.cels.2019.05.011
- ↵Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137. 10.1186/gb-2008-9-9-r137
- ↵Zheng H, Xie W. 2019. The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol 20: 535–550. 10.1038/s41580-019-0132-4