
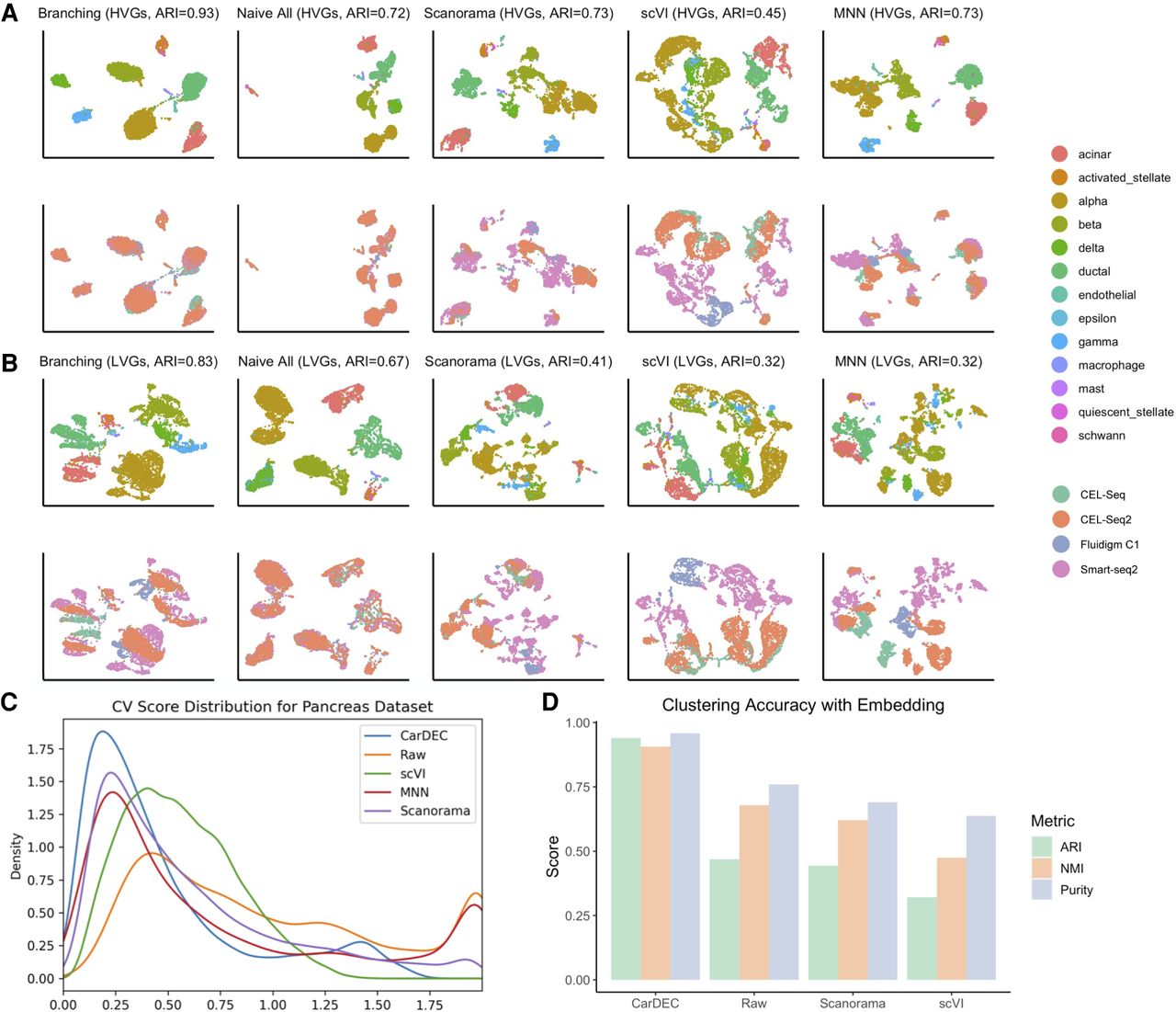
Justification for the branching architecture in CarDEC. The CarDEC API splits the input matrix into HVGs and LVGs and treats them separately with a “Branching” architecture as in Figure 1. Alternatively, we can use a “Naive” model that treats all features the same regardless of gene expression variance. The naive model consists of an autoencoder with a clustering loss in addition to the reconstruction loss. Here, we show the utility of the branching architecture. The HVGs and LVGs were clustered separately, and the ARI of assignments is provided along with a UMAP plot. The top row was colored by cell type, the bottom row by scRNA-seq protocol. (A) UMAP embedding computed from the denoised HVG counts for each method: CarDEC with Branching architecture, CarDEC with Naive Architecture, Scanorama, scVI, and MNN. (B) UMAP embedding computed from the denoised LVG counts for each method. Figure legends are the same as those in A. (C) Density plot of genewise coefficient of variation (CV) among batch centroids. Density plots are provided for HVGs and LVGs separately. (D) Clustering accuracy metrics were obtained using the embedding-based methods to cluster the data, rather than running Louvain on the full gene expression space. Results for “Raw” were obtained by using Louvain's algorithm on the original HVG counts and are provided as a baseline to which embedding-based clustering results may be compared.








