
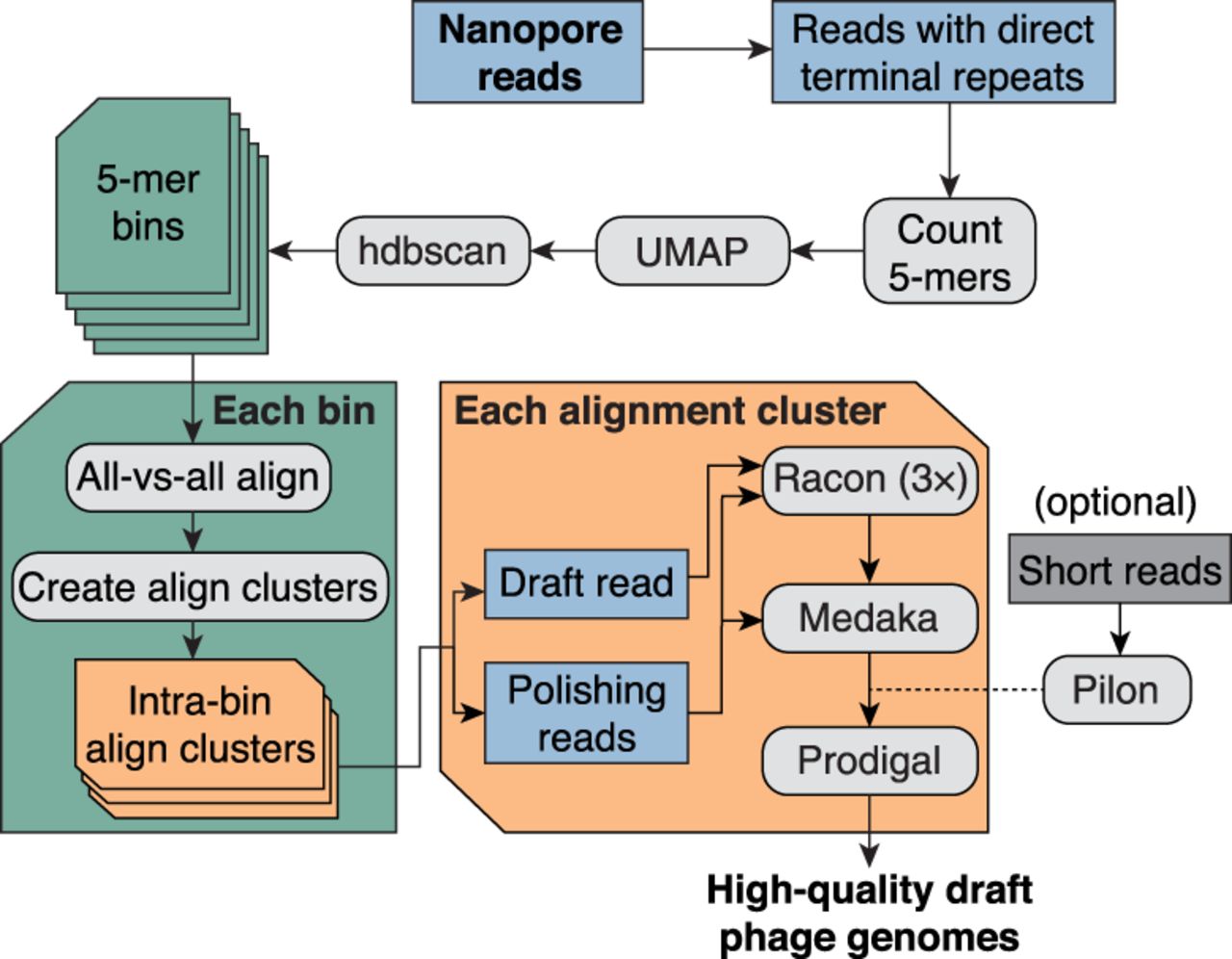
Bioinformatic pipeline for assembly-free discovery of marine phage genomes in nanopore sequences. Nanopore reads from each depth sample were first filtered to identify reads containing a direct terminal repeat (DTR). Read sequences were then decomposed into vectors containing their 5-mer counts, and all read vectors were embedded in a two-dimensional (2D) map using the dimensionality reduction tool UMAP (McInnes et al. 2018). Bins were called from the embedding using hdbscan (McInnes et al. 2017). Each 5-mer bin was refined by using minimap2 (Li 2018) to generate all-versus-all alignments of binned reads. Each pairwise alignment within each bin was assigned a score based on alignment length and sequence identity, and then these scores were hierarchically clustered to form one or more refined alignment clusters per 5-mer bin. Finally, a representative read was selected from each alignment cluster and polished by the remaining reads in the cluster using a combination of consensus polishing tools. Optionally, sample-matched short reads can be used to perform one last polishing round of the nanopore-only polished consensus sequence. Each polished draft genome was subsequently annotated for protein-coding sequences using Prodigal (Hyatt et al. 2010).








