
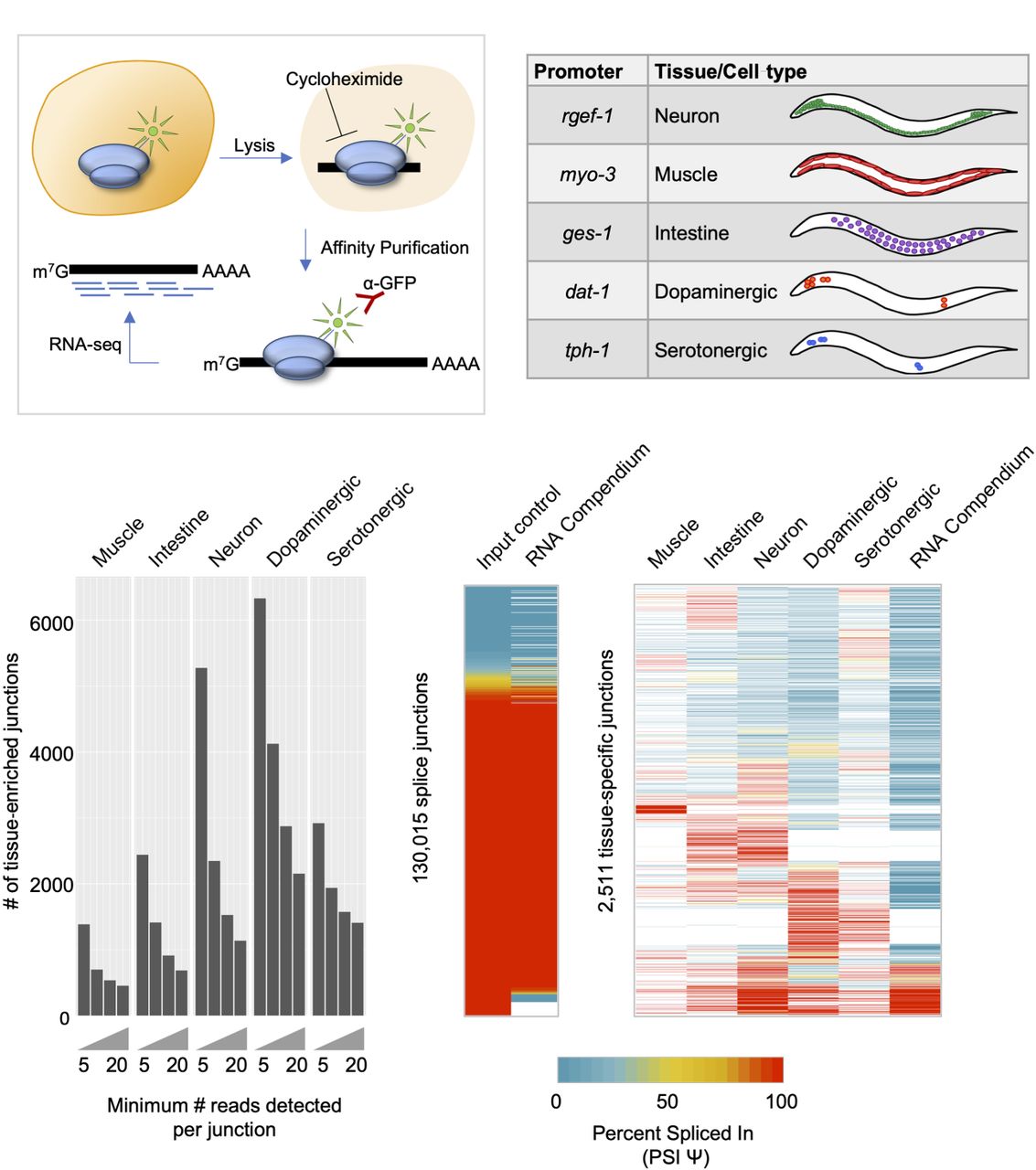
TRAP-seq identifies tissue-enriched junctions not found in whole-animal data. (A) Experimental outline for TRAP-seq approach. Animals expressing a GFP-tagged ribosomal protein are lysed in the presence of cycloheximide, and lysates are immunoprecipitated with anti-GFP antibodies, enriching for ribosome-associated mRNAs. cDNA libraries are prepared and then sequenced. (B) TRAP strains used in the present study, with reference gene listed from where promoters were cloned, as well as broad tissues or neuron subtypes for which mRNA profiles were obtained using TRAP-seq. (C) Number of tissue-specific splice junctions from each TRAP-seq IP sample that are absent from our whole-animal input control transcriptomes over increasing read count support thresholds. (D, left) Comparison of our whole-animal input control transcriptome data and RNA compendium data curated by Tourasse et al. (2017). PSI values for all input junctions and corresponding RNA compendium junctions are shown in the heatmap. Pearson correlation coefficient, R = 0.91, P < 2.2 × 10−16. (Right) PSI values of alternatively spliced tissue-specific junctions with at least 20 supporting reads, and corresponding PSI values in the RNA compendium. White indicates cases in which no reads were detected in a particular sample.








