
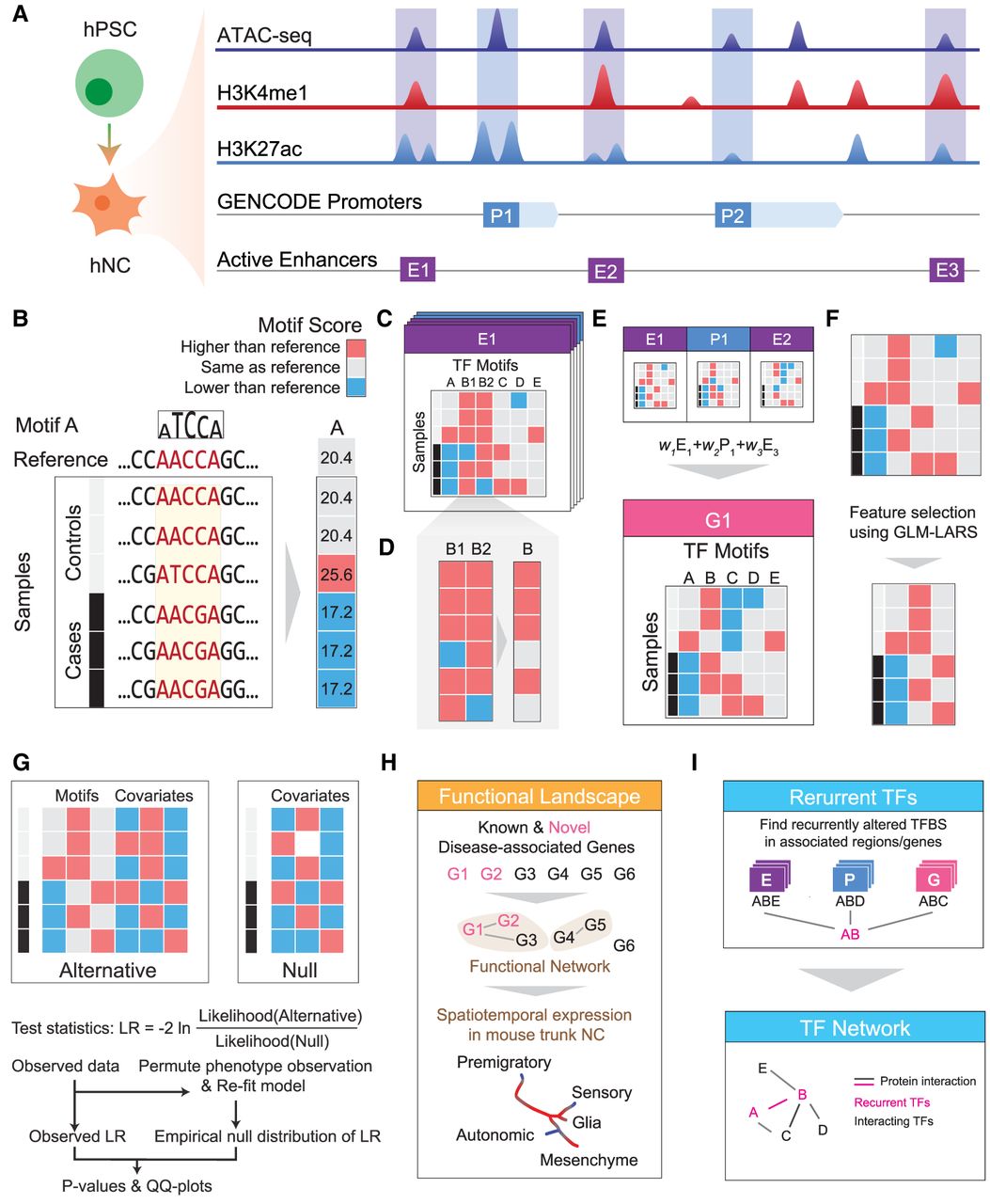
Schematic overview of MARVEL. (A) Epigenomic data of relevant cell types (hNC in the case of HSCR) are integrated with a gene annotation set to identify the active regulatory elements relevant to the phenotype of interest. (B) In each regulatory element, the functional significance of genetic variants is evaluated by their perturbation to TF sequence motifs. (C) Since the perturbation effects of multiple genetic variants may not add up linearly, they are considered together to reconstruct the sample-specific sequences, based on which the overall change of TF motif match scores is determined. (D) For motifs with multiple appearances within the same regulatory element, their match scores are aggregated to give a single score. (E) At a higher level, if a gene involves multiple regulatory elements, the aggregated match scores of a motif in the different elements can be further aggregated into a single score. This is done in the gene-based analysis. (F,G) The aggregated match score matrix of all the motifs for a regulatory element/gene is used as the input of an association test, which selects a subset of the most informative motif features (F) and compares a model involving both these selected features and the covariates with a null model that involves only the covariates using likelihood ratio (LR) test (G). (H) The regulatory elements and genes identified to be significantly associated with the phenotype can be further studied by other downstream analyses, such as gene set enrichment and single-cell expression analyses. (I) TFs with recurrently perturbed match scores in different regulatory elements are collected to infer a network that highlights the phenotype-associated perturbations.








