
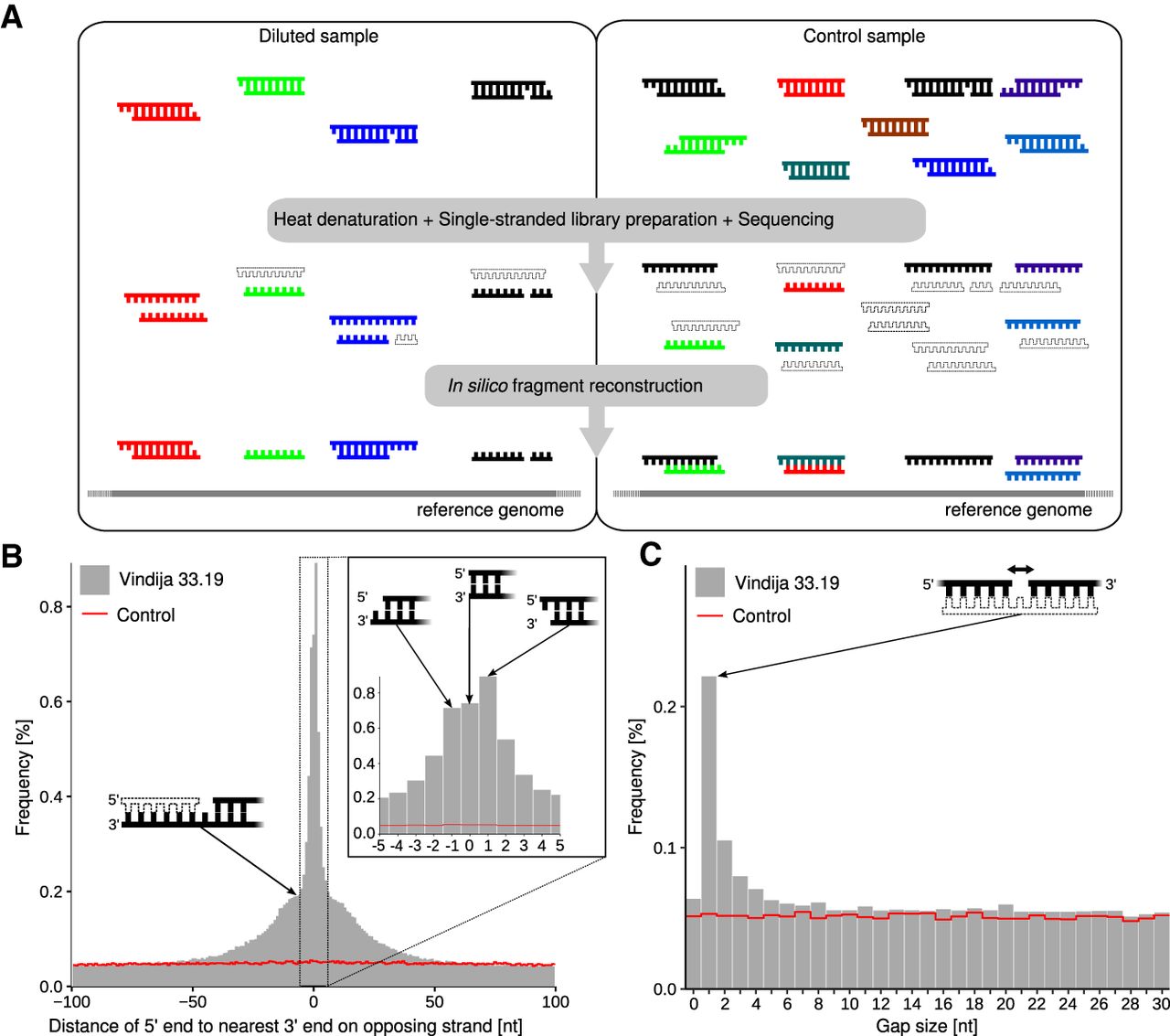
Identification of sequences from DNA strands originating from the same double-stranded DNA molecules. (A) Schematic overview of MatchSeq: DNA fragments isolated from ancient or recent biological material are diluted to a relatively small number, denatured by heat, converted into a single-stranded library, amplified, and deeply sequenced. Double-stranded fragments are then reconstructed in silico by identifying overlapping sequences mapping to the reference genome in reverse complementary orientation or in close proximity on the same strand. Sequences <35 bp cannot be confidently mapped to the reference and are discarded. The same analysis is performed for an equal number of control sequences obtained from shallow sequencing of a vast amount of artificially fragmented modern human DNA, in which sequences are expected to be distributed randomly across the reference genome. (B) Distribution of distances between the 5′ end of each sequence to the nearest 3′ end on the opposing strand obtained from Neanderthal DNA (Vindija 33.19; gray bars) and the control (red line). (C) Distribution of distances between sequence alignments on the same strand. Molecular structures putatively underlying the observed signals are indicated by schematic drawings. Filled DNA strands indicate sequences that were recovered; unfilled strands are hypothesized to be present but were not sequenced.








