
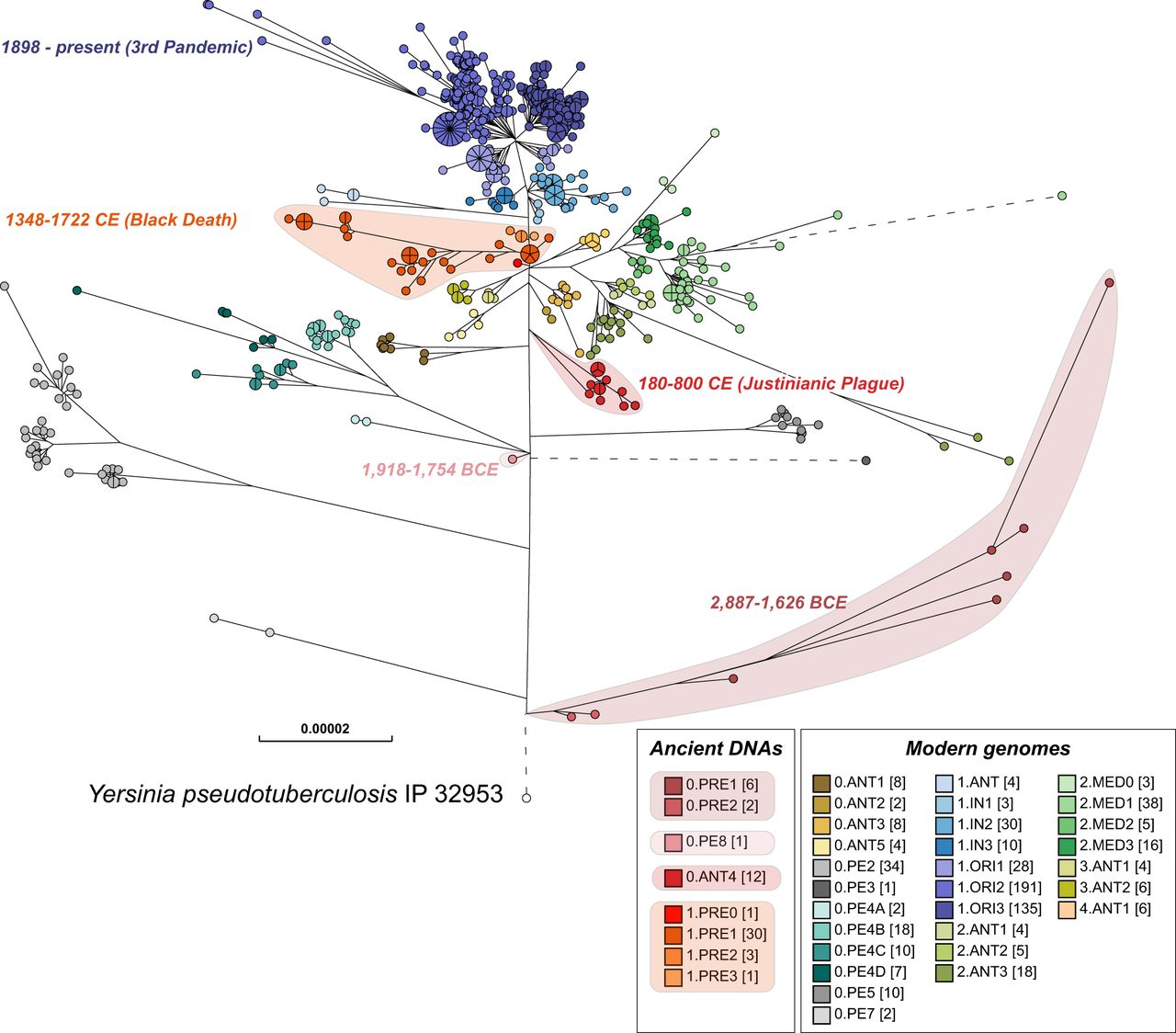
Maximum-likelihood tree of modern and ancient genomes of Y. pestis. EnteroBase contained 1368 ancient and modern Y. pestis genomes in October 2019, of which several hundred genomes that had been isolated in Madagascar and Brazil over short time periods showed very low levels of genomic diversity. To reduce this sample bias, the data set used for analysis included only one random representative from each HC0 group from those two countries, leaving a total of 622 modern Y. pestis genomes. Fifty-six ancient genomes of Y. pestis from existing publications were assembled with EToKi (Methods), resulting in a total of 678 Y. pestis genomes plus Yersinia pseudotuberculosis IP32953 as an outgroup (http://enterobase.warwick.ac.uk/a/21975). The EnteroBase pipelines (Supplemental Fig. S2D) were used to create a SNP project in which all genomes were aligned against CO92 (2001) using LASTAL. The SNP project identified 23,134 nonrepetitive SNPs plus 7534 short inserts/deletions over 3.8 Mbps of core genomic sites which had been called in ≥95% of the genomes. In this figure, nodes are color coded by population designations for Y. pestis according to published sources (Morelli et al. 2010; Cui et al. 2013; Achtman 2016), except for 0.PE8 which was assigned to a genome from 1918 to 1754 BCE (Spyrou et al. 2018). The designation 0.ANT4 was applied by Achtman (2016) to Y. pestis from the Justinianic plague described by Wagner et al. (2014), and that designation was also used for a genome associated with the Justinianic plague (DA101) that was later described by Damgaard et al. (2018) as 0.PE5.








