
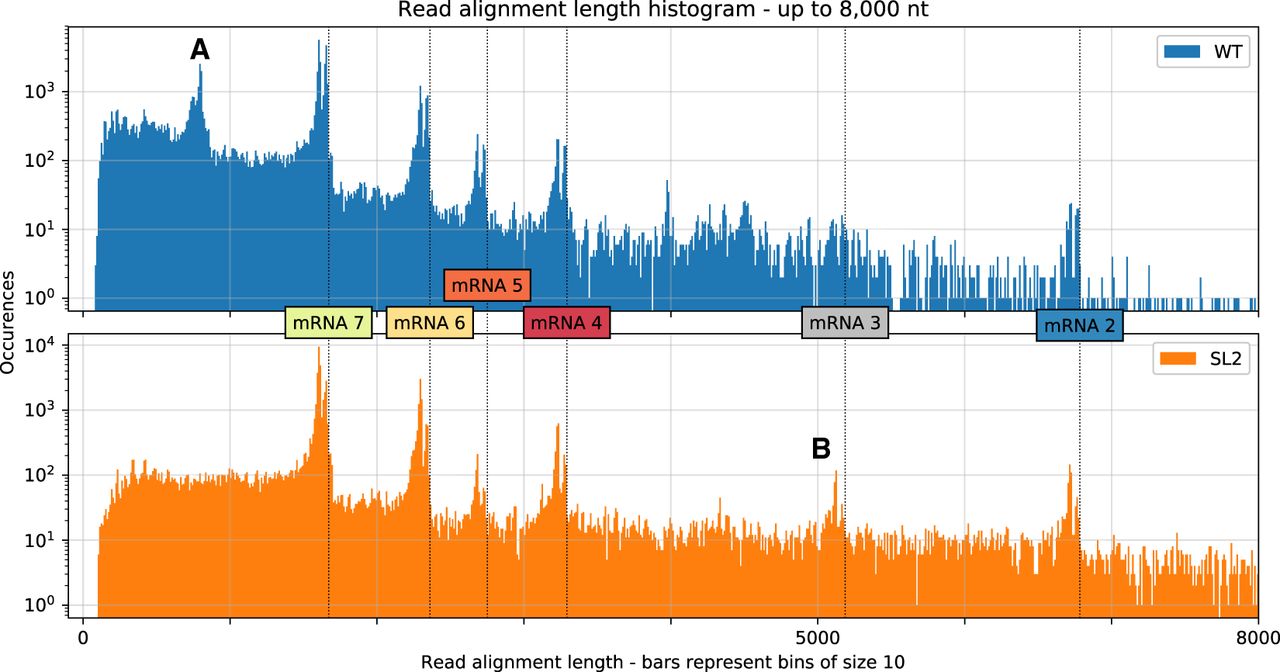
Distribution of aligned read lengths up to 8000 nt for both samples based on alignments with minimap2. We observed clusters that correspond well with the transcript lengths expected for canonical mRNAs (vertical dotted lines). Alignment of the reads to these canonical mRNA sequences confirmed these observed abundances (Supplemental Fig. S5). The distribution shows double peaks at the cluster positions because reads corresponding to mRNAs often miss the leader sequence, possibly owing to basecalling or mapping errors. We also observed additional clusters that likely correspond to highly abundant DI-RNAs. (A) A cluster in the WT sample that represents chimeric ∼820-nt sg RNAs composed of both 5′- and 3′-terminal genome regions (∼540 and ∼280 nt, respectively). We propose the template switch for this transcript occurs around position 27,000 and RNA synthesis resumes at around position 540 (see corresponding peaks in Fig. 3, track B). (B) A cluster from the SL2 sample that contains transcripts with an approximate length of 5150 nt, which represents mRNA 3 (see Fig. 1). These transcripts are probably formed owing to a transcription stop at a TRS motif around position 22,150 (see corresponding peak in Fig. 3, track C).








