
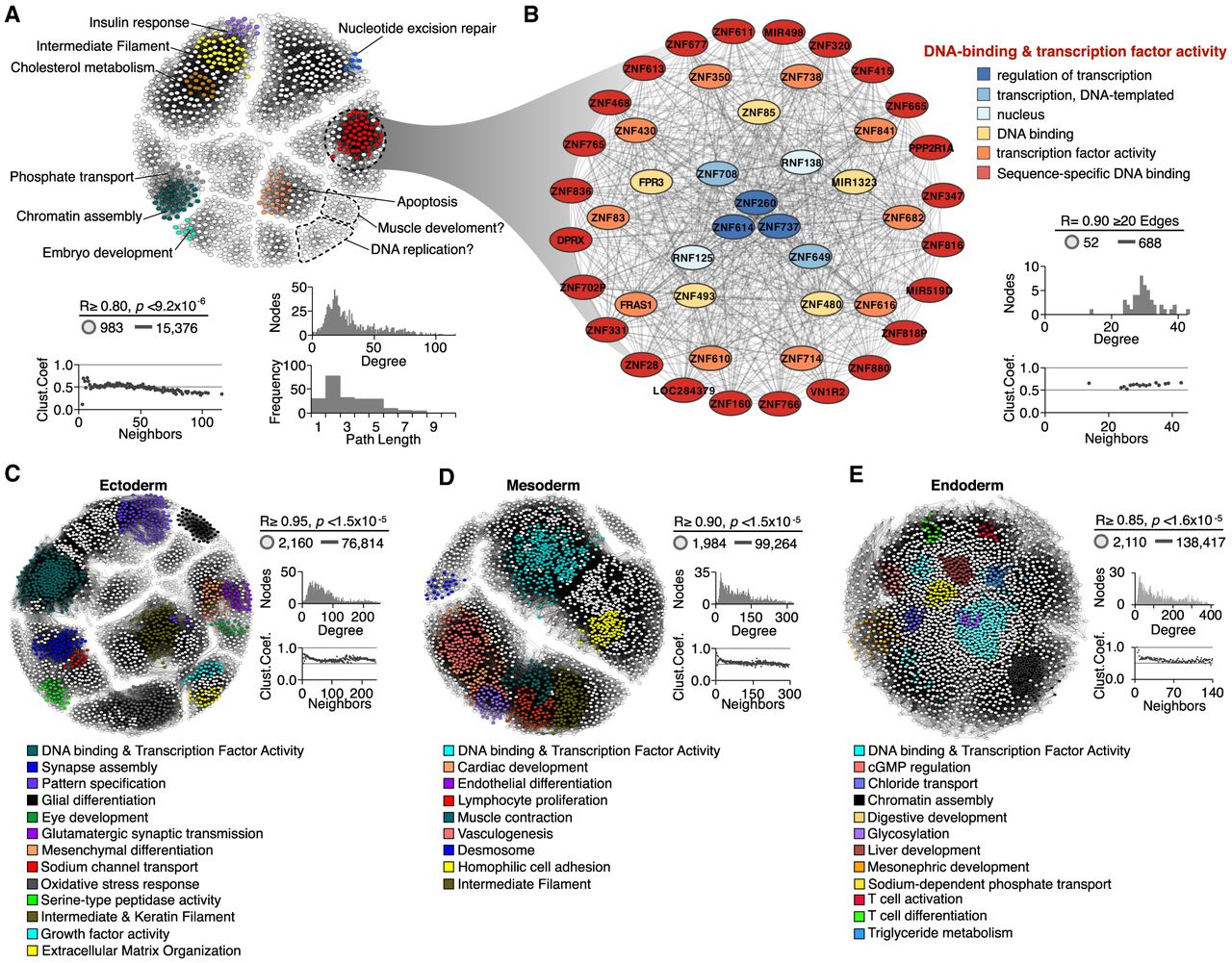
Functional annotation of RT networks. (A) RT-correlated gene pairs across all differentiation pathways from human ES cells were identified and exploited to construct a RT network. (B) Detailed subnetwork connectivity from the RT network shown in A and its respective node organization per ontology term. (C–E) RT networks and functional subnetwork communities constructed for each differentiation pathway: ectoderm (C), mesoderm (D), and endoderm (E). Interaction edges between gene pairs were established only for significantly correlated nodes (Bonferroni's adjusted P-values with alpha = 0.05/n), and the subset of most connected nodes (>20 edges) was used to visualize RT networks displayed as 2D maps in Cytoscape (Shannon et al. 2003). Pearson's correlation and Bonferroni-corrected P-value thresholds, as well as the connectivity analysis for each network, are shown (degree distribution, path lengths, and clustering coefficients). Highly interconnected subnetwork communities were annotated with functional ontology terms using the SAFE algorithm (Baryshnikova 2016) and are displayed in distinct colors.








