
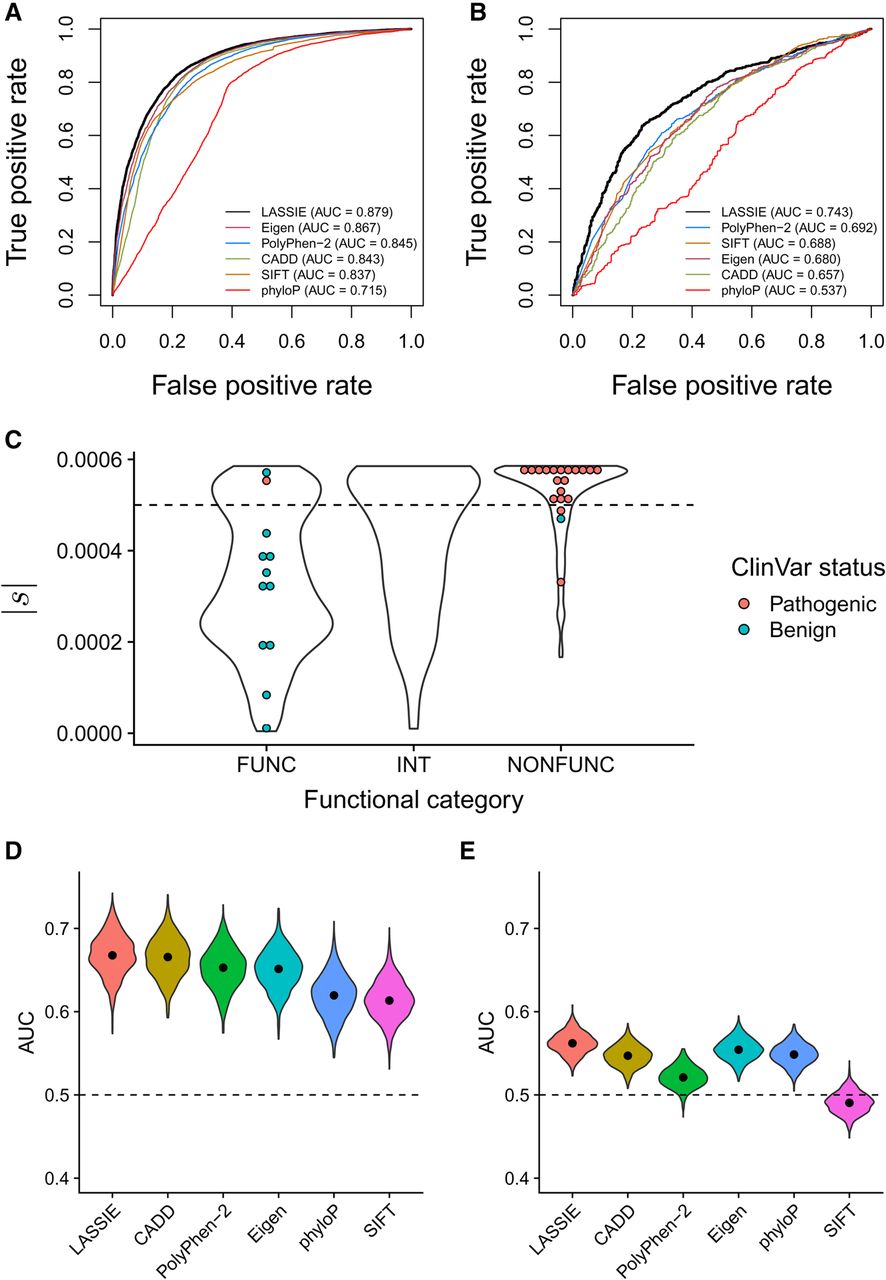
Performance in predicting disease-associated nonsynonymous variants. Performance is quantified using the area under the receiver operating characteristic curve (AUC) statistic. Results for LASSIE are compared with those for Eigen (Ionita-Laza et al. 2016), PolyPhen-2 (Adzhubei et al. 2010), CADD (Kircher et al. 2014), SIFT (Ng and Henikoff 2003), and phyloP (Pollard et al. 2010). (A) Performance for pathogenic variants from ClinVar (Landrum et al. 2014). (B) Performance for cancer-driver mutations from Chang et al. (2016). (C) Distributions of estimated |s| for variants in BRCA1 predicted to be “functional” (FUNC; i.e., nondisruptive), “intermediate” (INT), or “nonfunctional” (NONFUNC; i.e., disruptive) by saturation genome editing (Findlay et al. 2018). Colored dots indicate those variants also having expert-reviewed status in ClinVar (CLINREVSTAT = reviewed by expert panel). (D) Performance for rare (MAF < 1%) GWAS hits. (E) Performance for common (MAF > 5%) GWAS hits, showing that all methods have limited power.








