
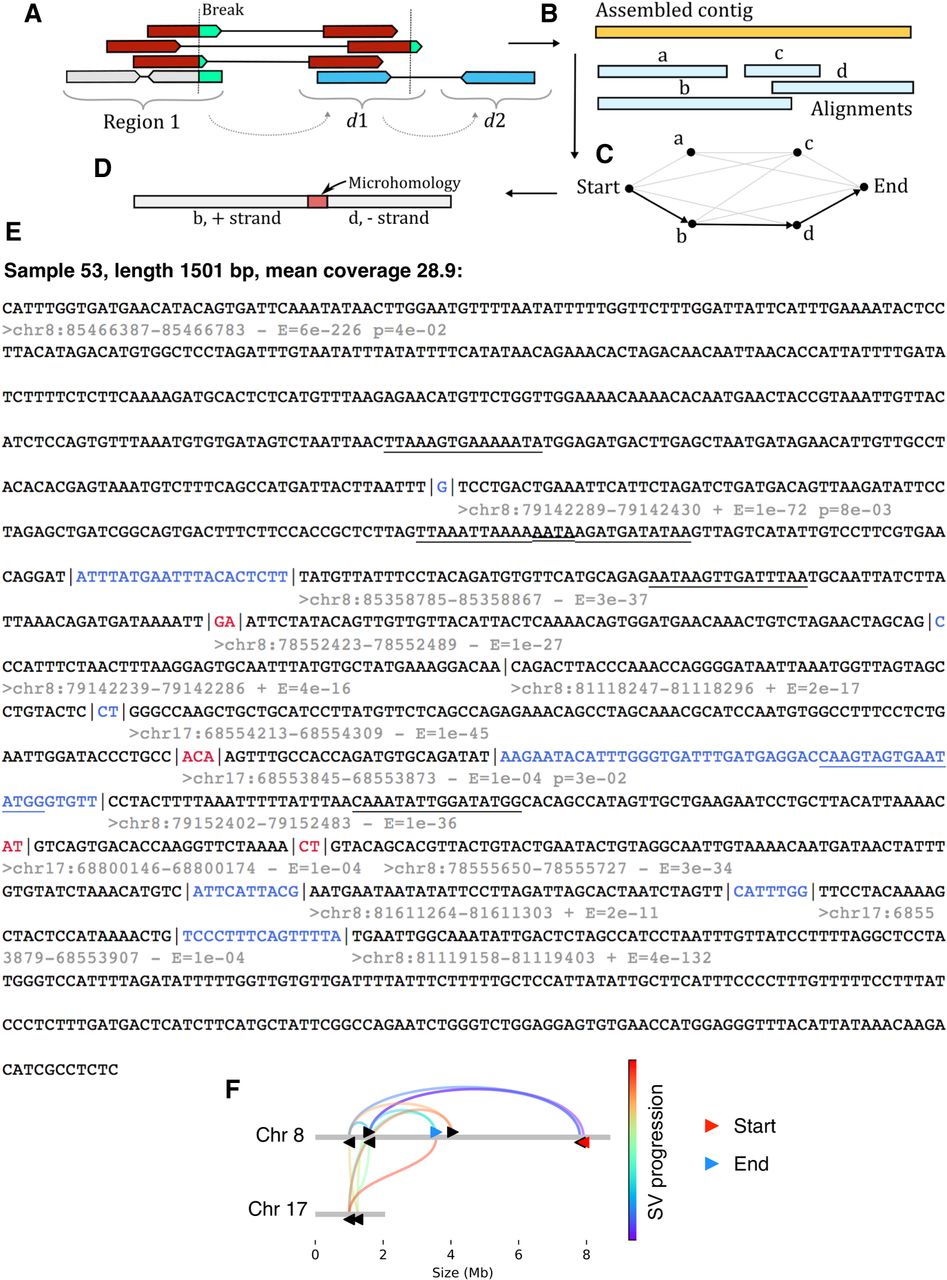
Assembly of complex SVs reveals signatures of replicative repair. (A) Reads were collected from breakpoint regions before recursively fetching mate pairs from distal genomic loci. (B) Reads were then assembled into contigs and aligned to the reference genome. An optimal set of alignments was then determined using the fnfi align algorithm https://github.com/kcleal/fnfi (C) before further annotation of the contigs (D). (E) An example contig from LIG3−/−:NC3:sample 53 is provided with the following annotations: Black text demarcates alignment to the reference genome, whereas red and blue indicate sections of microhomology and novel sequence insertions, respectively. Below the start of each alignment, the genomic interval is given along with the DNA strand and an E-value for the alignment. An additional annotation is added to lines for which adjacent alignments show significant levels of similarity with respect to one another, as determined by a statistical test, giving the probability “p” indicated in the annotation. Significant stretches of similarity have been underlined in adjacent segments and overlapping annotations appear as a double underlined stretch. (F) The progression of rearrangement over the reference genome is depicted.








