
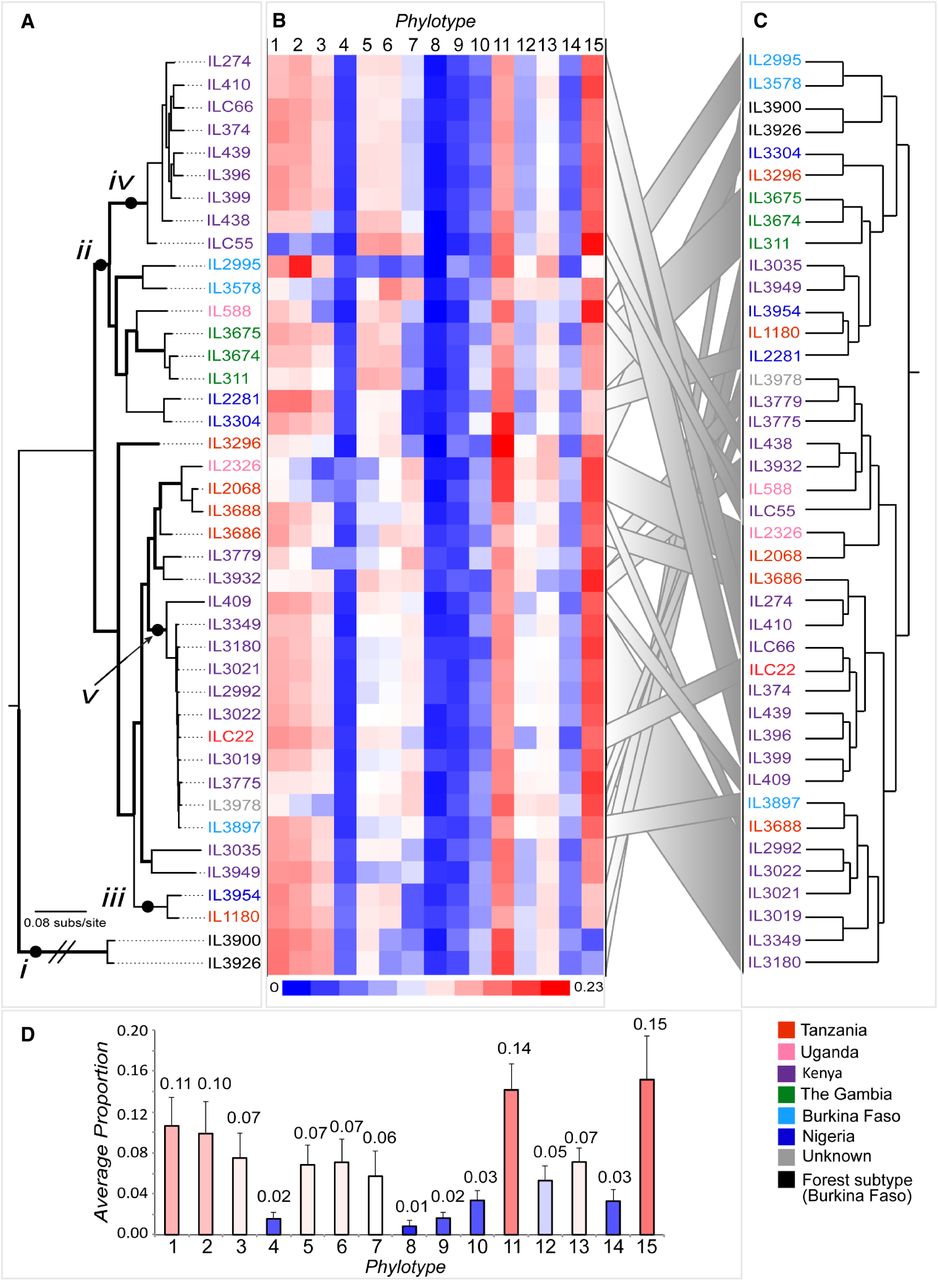
Relationships between the VSG repertoire, geography, and population structure in T. congolense. (A) ML phylogeny of T. congolense strains in this study based on whole-genome single-nucleotide polymorphisms (SNPs), estimated with RAxML (Stamatakis 2014) with a GTR + Γ model and 100 bootstrap replicates (branches with bootstrap greater than 70 are shown in bold). Labels “i” to “v” denote examples referred to in the text. Label “i” shows the long phylogenetic distance between T. congolense Savannah and Forest subtypes; “ii” points to the only clade maintaining a geographic signature. Labels “iii,” “iv,” and “v” show examples of lack of concordance between the population history recapitulated by the SNP phylogeny and the VAP, demonstrated by the dendrogram. (B) VAPs for all strains shown as a heatmap of the proportions of 15 universal phylotypes. (C) A dendrogram depicting the relationships among VAPs based on Euclidian distances estimated in R. Gray ribbons link the position of parasite strains in A and C. (D) A bar chart showing the average proportion of each phylotype (mean ± σ) across all strains. Strains are color-coded by provenance according to the key.








