
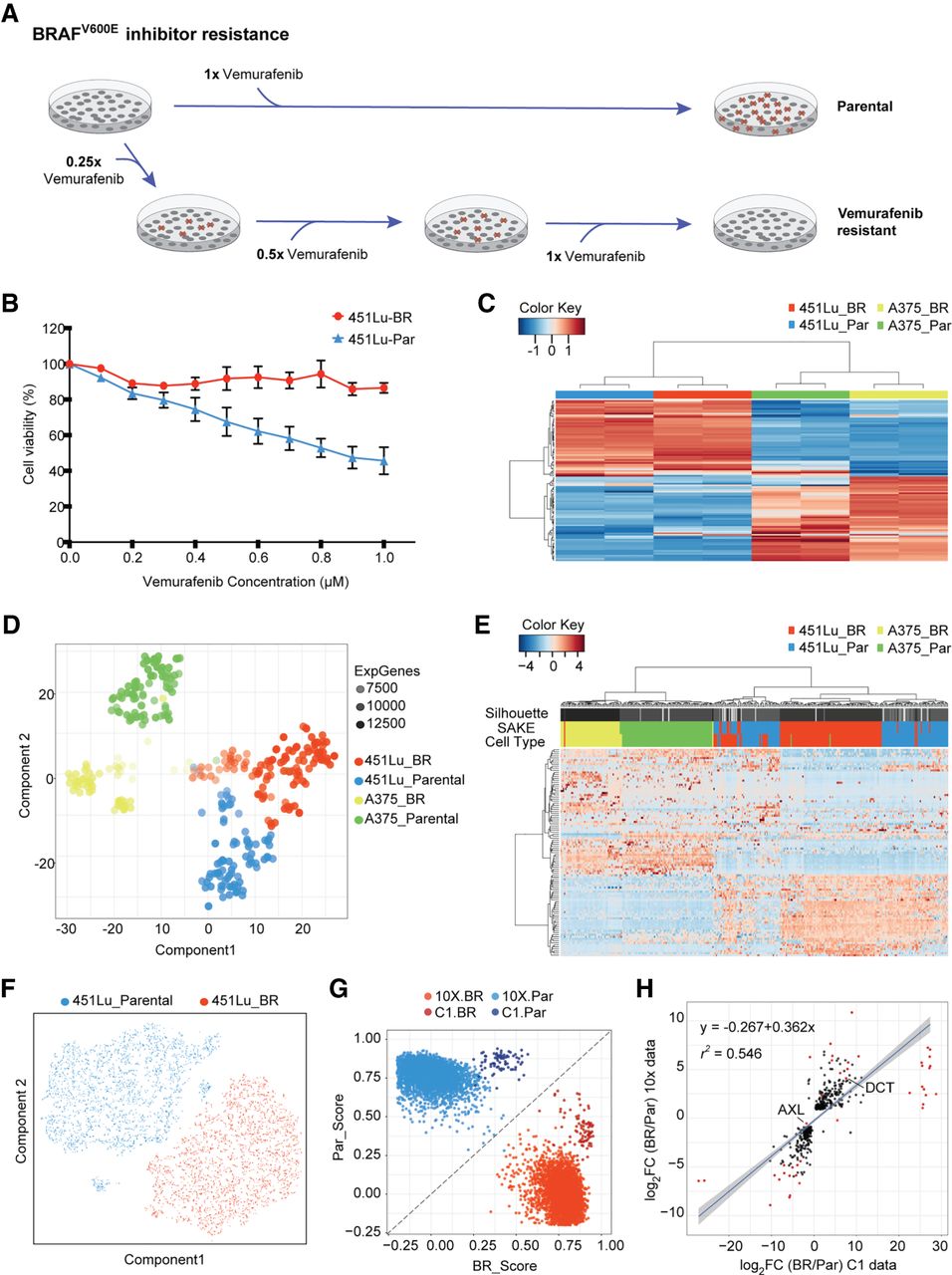
Bulk and single-cell RNA-seq were used to study differential drug responses to BRAF inhibitor treatment. (A) Naïve melanoma cells were treated with an increasing dose of the BRAF inhibitor, vemurafenib. Cells that survived after each drug treatment were selected to gradually derive stably BRAFi-resistant cells. (B) Drug sensitivity was measured through the use of MTT assays to assess metabolically active cells 72 h following BRAFi application. (C) Melanoma signature gene sets (Hoek et al. 2008) were used to cluster bulk RNA-seq data from melanoma cell lines. (D) A t-SNE map was used to display the expression profiles from approximately 400 parental and BRAFi-resistant melanoma cells isolated using the Fluidigm C1 platform. The first t-SNE component separates the two cell lines, and the second component distinguishes between parental and BRAFi-resistant cells. (E) Highly expressed and variable genes were used to classify Fluidigm C1 scRNA-seq data. Higher levels of heterogeneity can be observed among 451Lu cells as compared to A375 cells. (F) To determine whether 451Lu cells have more intrinsic heterogeneity, 6545 scRNA-seq transcriptomes were obtained using the 10x Chromium platform. A t-SNE map of this 451Lu 10x data highlights two major groups of cells: parental (blue) and BRAFi resistant (red). (G) In order to compare the 10x and C1 data on the same scale, a scoring system was implemented to determine the Spearman's rank correlation distance of each cell from the centroids of the parental (PAR) and BRAFi-resistant (BR) populations. (H) Differential expression analysis identified genes significantly altered in 451Lu-BR versus 451Lu-Par cells, with log2 fold change values plotted for the C1 data set (horizontal axis) and 10x data set (vertical axis). Genes that have adjusted P < 1 × 10−50 were highlighted in red, with all statistically significant genes (P < 0.01) shown in black, showing highly similar results.








