
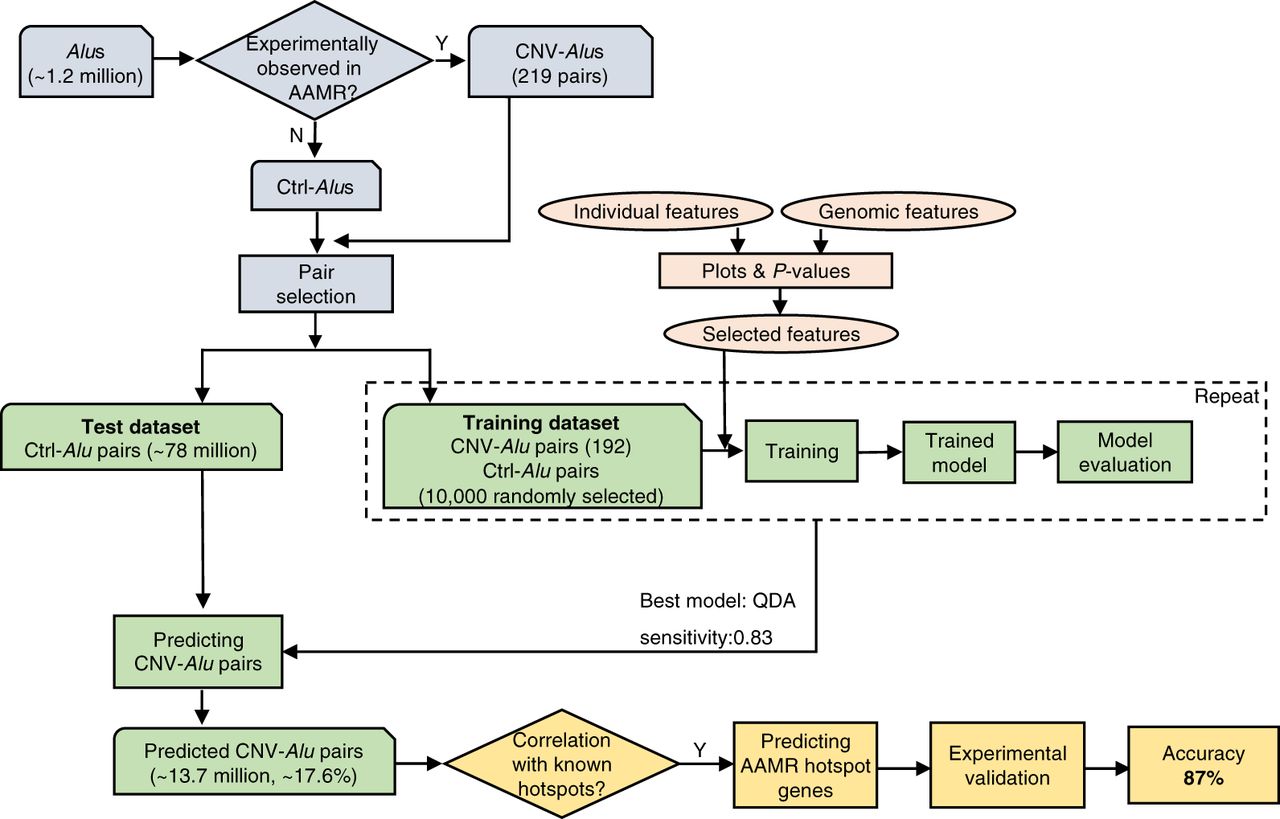
Diagram of the workflow used for predicting CNV-Alu pairs and AAMR hotspot genes in this study. Approximately 1.2 million Alus are documented in the “Repeating Elements by RepeatMasker” track at the UCSC Genome Browser. CNV-Alus are those with experimental evidence supporting their role in AAMR (Supplemental Table S1), and all the others are Ctrl-Alus. We selected Alu pairs that are in the same orientation, span at least one exon, and are located <250 kb from each other. Both the individual Alu sequence features and genomic architectural features were characterized, and a subset of features were utilized in model training. The QDA (quadratic discriminant analysis) model achieved the highest sensitivity and was applied for predicting CNV-Alu pairs. The amount of predicted CNV-Alu pairs is significantly correlated with the number of observed AAMR events for known hotspot genes. Therefore, we further determined the relative risk of AAMR in 12,074 human genes that have a MIM entry using the count of predicted CNV-Alu pairs. Finally, we experimentally validated this prediction with 89 samples selected by correlating predicted hotspot genes with a database of approximately 54,000 chromosomal microarrays (CMAs) by performing aCGH and mapping the breakpoint junctions of detected CNVs. We achieved an 87% positive predictive value overall.








