
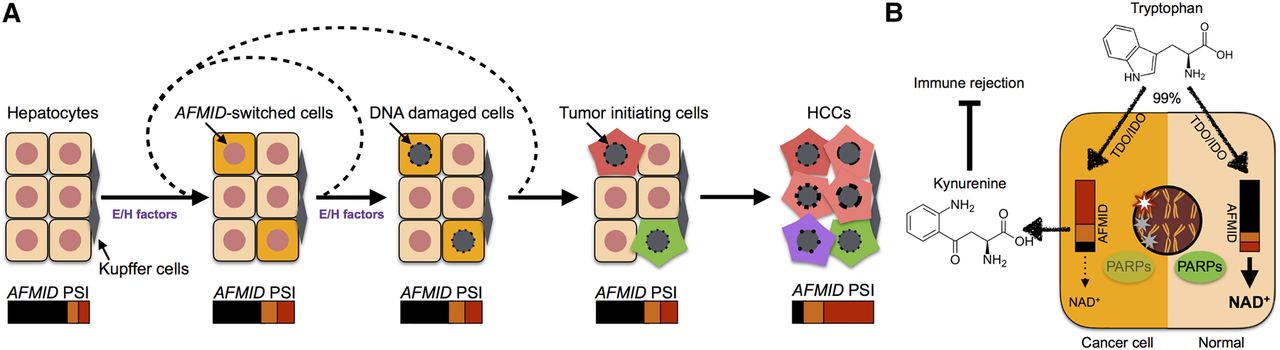
Model of the AFMID isoform switch in HCC. (A) The flow chart represents HCC progression (left to right). At the start, six representative hepatocytes are shown (nuclei in red). Later, because of environmental or hereditary factors (E/H factors), a subset of hepatocytes switches AFMID isoforms. The environmental factors include WNT signals in peri-central hepatocytes in daily liver regeneration, cytokines released during inflammation, chemical damage, and virus infection. The hereditary factors include driver mutations, such as in TP53 and ARID1A. The E/H factors temporarily disrupt the identity of hepatocytes and reduce the NAD+ level in the hepatocytes. The reduced NAD+ level gives rise to increased DNA damage in the nucleus. After recursively accumulating driver mutations, the DNA-damaged hepatocytes become HCC initiating cells. The switch of AFMID isoforms can then be observed by bulk RNA-seq. (B) The diagram shows two states of hepatocyte cells. On the right, a normal hepatocyte expresses high levels of AFMIDFL isoform, and tryptophan can be converted to NAD+ for PARPs to fix DNA damage. On the left, the HCC cell expresses low levels of AFMIDFL and has low NAD+, so DNA damage is increased and kynurenine is secreted to inhibit immune rejection.








