
Abstract
We present a method to detect copy number variants (CNVs) that are differentially present between two groups of sequenced samples. We use a finite-state transducer where the emitted read depth is conditioned on the mappability and GC-content of all reads that occur at a given base position. In this model, the read depth within a region is a mixture of binomials, which in simulations matches the read depth more closely than the often-used negative binomial distribution. The method analyzes all samples simultaneously, preserving uncertainty as to the breakpoints and magnitude of CNVs present in an individual when it identifies CNVs differentially present between the two groups. We apply this method to identify CNVs that are recurrently associated with postglacial adaptation of marine threespine stickleback (Gasterosteus aculeatus) to freshwater. We identify 6664 regions of the stickleback genome, totaling 1.7 Mbp, which show consistent copy number differences between marine and freshwater populations. These deletions and duplications affect both protein-coding genes and cis-regulatory elements, including a noncoding intronic telencephalon enhancer of DCHS1. The functions of the genes near or included within the 6664 CNVs are enriched for immunity and muscle development, as well as head and limb morphology. Although freshwater stickleback have repeatedly evolved from marine populations, we show that freshwater stickleback also act as reservoirs for ancient ancestral sequences that are highly conserved among distantly related teleosts, but largely missing from marine stickleback due to recent selective sweeps in marine populations.
Comparing closely related vertebrate species, such as humans and chimpanzees, reveals that duplications and deletions are the classes of mutations that have affected the greatest number of base pairs (Cheng et al. 2005). Duplications and deletions underlie copy number variation, which occurs when a genomic segment appears a variable number of times in different species or in individuals of the same species.
Copy number variation is not only common (Sharp et al. 2005; Chain et al. 2014), but also capable of producing large phenotypic effects. Many of these effects are deleterious, and both increases and decreases in copy number are associated with human diseases. For example, duplications are associated with Charcot–Marie–Tooth disease (Lupski et al. 1991) and deletions with cri-du-chat syndrome (Punnett et al. 1964). Somatic copy number changes, such as those harboring ERBB2 and BRCA1, are also associated with cancer (Savinainen et al. 2002; Birgisdottir et al. 2006).
However, some copy number variants are advantageous. Greater copy number of the amylase gene has been found in human populations that eat high-starch diets (Perry et al. 2007), and more copies of the CCL3L1 gene are associated with decreased susceptibility to HIV/AIDS (Gonzalez et al. 2005). Deletions have also been shown to be adaptive. For example, stickleback fish have repeatedly lost their pelvic apparatus in particular environments (Bell et al. 1993). The causative mutations are recurrent deletions of a pelvic enhancer near the PITX1 gene, and these deletion alleles show molecular signatures of positive selection in freshwater populations (Chan et al. 2010). Since the split with chimpanzees, humans have deleted enhancers near the androgen receptor gene AR, the bone morphogenetic protein gene GDF6, and the tumor-suppressor gene GADD45G, which have likely contributed to the loss of penile spines, the modification of digits, and an increased brain size in humans (McLean et al. 2011; Indjeian et al. 2016).
Sanger sequencing allowed copy number to be inferred by analyzing read depth (Bailey et al. 2002), studying the spacing and orientation of paired reads (Tuzun et al. 2005), identifying reads that span junctions not present in the reference genome (Mills et al. 2006), or assembling reads and aligning the assembly to a reference genome (Levy et al. 2007). Second-generation sequencing technologies that produce relatively inexpensive short reads are now commonly used to identify copy number variation in samples (Mills et al. 2011). Most of these methods identify copy number variants in a single sample, often in relation to a reference genome for the species or a reference specific to an individual in the case of tumor and matched normal tissue (Chiang et al. 2009). Pairwise methods have also been developed that identify copy number differences between two samples when mapped to the same reference genome (Kim et al. 2010). A recent method uses a population-based approach which identifies CNVs in the pool of samples and then genotypes each individual for those CNVs (Handsaker et al. 2015).
We present a method to identify copy number variation that correlates with an ecological or phenotypic variable, which divides a set of samples into two groups. The method is in the form of a finite-state transducer. Although transducers have only recently seen increased use in biological applications (Bradley and Holmes 2007), finite-state machines, in the form of the hidden Markov model, have been widely used in biological sequence analysis for decades (Krogh et al. 1994; Burge and Karlin 1997; Eddy 1998). Compared to hidden Markov models, transducers allow for conditional probabilities.
To better understand the contribution of copy number variation to ecological adaptation in vertebrates, we applied this method to the genomes of 21 geographically diverse marine and freshwater threespine stickleback (Gasterosteus aculeatus) fish (Jones et al. 2012). The stickleback is a powerful model organism for understanding the molecular basis of vertebrate adaptation, because marine fish have repeatedly colonized a wide variety of freshwater habitats following the melting of glaciers, beginning approximately 10,000–20,000 yr ago (Bell and Foster 1994; Clark et al. 2009). Similar phenotypes have repeatedly evolved in similar ecological contexts, which suggests that the corresponding traits have been positively selected in the new environment. By identifying regions of the genome where the copy number at a locus correlates with an ecological variable, as opposed to the typical correlation with geography (Chain et al. 2014), we can discover copy number differences that are associated with recurrent adaptation to a particular environment. Previous studies in human genetics have also identified copy number variants that show strong differentiation between populations (Sudmant et al. 2015). Sticklebacks have the advantage that many replicate populations have evolved in similar environments, making it possible to test whether the same copy number variants show repeated differentiation among independent populations adapting to similar ecological conditions. In this paper, we contrast populations from marine and freshwater habitats as our ecological variable of interest, but the methods we have developed could be applied to any binary ecological variable or phenotypic trait.
Results
Data set
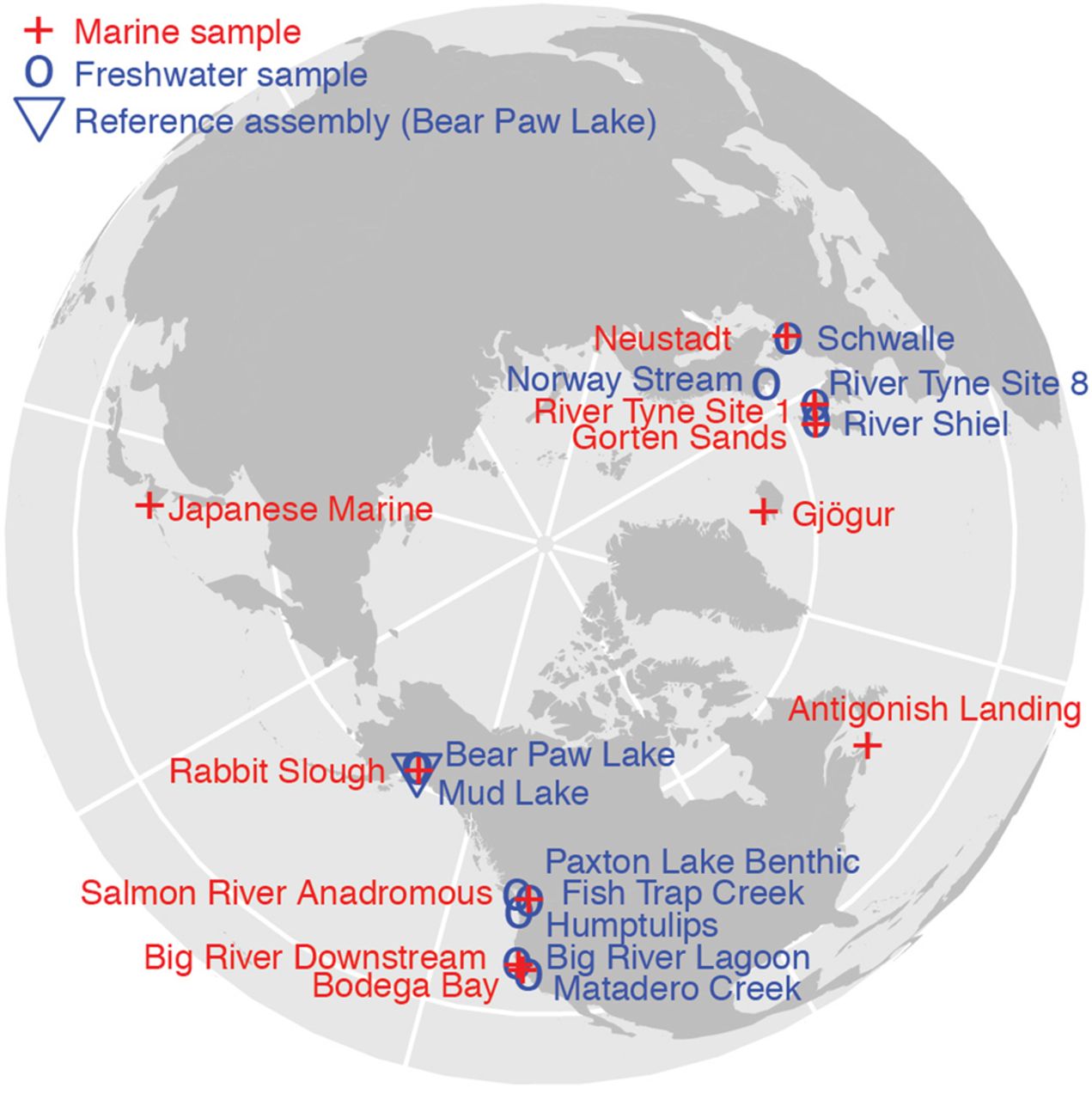
To detect copy number variation that repeatedly differentiates freshwater from marine stickleback populations, we used a data set of 21 whole-genome sequencing libraries from 11 freshwater and 10 marine stickleback fish that were collected from throughout the Northern Hemisphere (Fig. 1; Jones et al. 2012). The sequence reads are 36 bp in length, and the coverage ranges from 0.4× to 3.2× with a median of 1.7×.
The collection locations of the 11 freshwater stickleback (blue) and 10 marine stickleback (red). Of the 21 sites, 14 belong to marine–freshwater pairs in which the sampling sites are geographically adjacent and have likely experienced ongoing or historical gene flow. Such gene flow reduces the number of genetic differences that are not related to differing marine and freshwater habitats.
Model
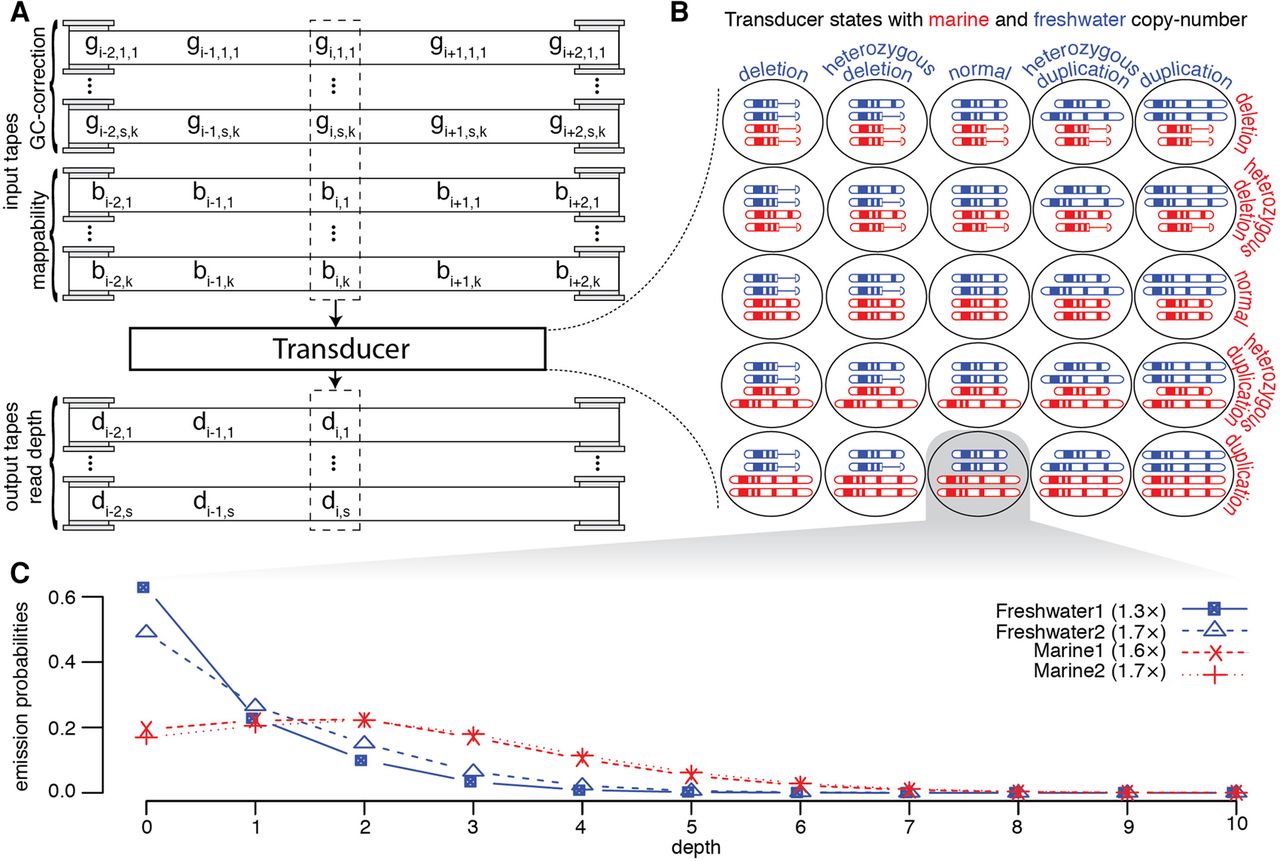
We used the framework of a finite-state transducer to identify locations in the genome where the resequencing data supports the canonical marine copy number being different from the canonical freshwater copy number. The finite-state transducer considers the following canonical copy numbers: homozygous deletion, heterozygous deletion, consistent with reference, heterozygous duplication, and homozygous duplication for freshwater genomes and marine genomes. The transducer has a state for all possible combinations of copy number for each ecological group, resulting in 25 states (Fig. 2).
The transducer model. (A) The transducer reads from s · k+k tapes containing the GC-bias and mappability of all overlapping reads, where s is the total number of individuals being analyzed, and k is the length of sequencing reads. gi,n,j is the GC-bias of the jth of k read position that would cover assembly position i for sample n of s. bi,j is the mappability of the jth of k read position that would cover position i. There are multiple output tapes, one for each individual, containing the read depth for that sample. di,n is the depth of reads at assembly position i for sample n of s. (B) The 25 states represent the variation in canonical copy number that can be detected by the transducer. (C) One example of the emission probabilities for a single base with the state representing no change in freshwater copy number, while the marine fish have a duplication. We show the marginal probability distribution for four individuals.
The transducer does not emit only one depth of coverage, but rather a tuple of coverages, one for each sample, thereby analyzing all samples simultaneously (Supplemental Methods). This enables the detection of copy number variation that is shared by many of the samples, even in cases in which there is insufficient power to detect copy number variation in an isolated individual. In addition, the ecologically differentiated deletions or duplications need only share regions that are gained or lost in common. The algorithm does not require the individual samples to share deletion and duplication breakpoints. This allows the method to find both the reuse of identical alleles selected from standing variants shared among populations, as well as the repeated duplication or deletion of a region through de novo mutations. The method is robust to assembly errors in the reference genome since it is identifying regions where the canonical marine and freshwater genomes differ, rather than identifying differences from the reference assembly.
Read depth emission probabilities for each sample are specific to each base pair in the genome. Previous methods used the binomial or Poisson distributions to model read depth, and when the observed distribution of read depths was found to be overdispersed, the negative binomial was used to better fit the data (Magi et al. 2012). We present the read depth over a genomic segment as a mixture of binomial distributions, in which each base has its own uniquely calculated probability of being covered by a read, based on the mappability and GC-content of the reads that could cover that base. Thus, the read depth over an interval of x bases is a mixture of x binomial distributions (Supplemental Methods). Deleted regions may not be completely devoid of reads, because some reads from elsewhere in the genome may mismap into the region. Duplicated regions may not fully double their number of reads, because some reads from the region may mismap to paralogous regions that were not duplicated. Based on the mappability of reads that could cover a base, we estimated how likely a base is to be covered by a read even when deleted and how likely a base is to lose additional read depth to other places in the genome when duplicated (Methods). We also modeled the GC-bias of sequencing libraries (Ross et al. 2013) at base-level for each sample (Methods). This statistical framework not only better captures the overdispersion previously observed when the coverage of all bases in a genomic region are pooled together, but it also tailors the expected read depth for each base position instead of using the same emission probabilities for all genomic positions. The improvement is minor in duplications, but substantial in deletions (Supplemental Figs. S1, S2).
We computed the most likely series of hidden states that would produce the observed read depth, given the GC-content and mappability as inputs to the transducer. This decoded series of hidden states represents the canonical copy number of samples in group one (freshwater) and the canonical copy number of samples in group two (marine) at each genomic position. States in which the canonical copy numbers for the two sample groups are not equal identify regions of the genome where copy number variation correlates with group membership (marine or freshwater environment).
Performance on simulated data sets
To evaluate the performance of the transducer, we simulated data sets that are analogous to the sequences collected from wild fish, but with known introduced regions of copy number variation between the marine and freshwater ecotypes (Supplemental Methods). We optimized the transducer's transition probabilities to detect true differences, while limiting the expected number of false positives to 0.2 (Supplemental Methods).
We detected the majority of randomly placed simulated deletions down to <40 bp and duplications down to ∼250 bp, when these mutations are present in all members of one group, while the other group is consistent with the reference genome (Fig. 3; Supplemental Figs. S3, S4). To put these results in perspective, we repeated the analysis using previously available approaches to detect repeated deletions or duplications that correlate with membership in one of two groups. CNVnator (Abyzov et al. 2011), rSW-seq (Kim et al. 2010), cn.MOPS (Klambauer et al. 2012), and Genome STRiP (Handsaker et al. 2015) are widely used programs for detecting copy number variation from short sequencing reads. Although these programs do not explicitly annotate CNVs that differentiate two groups of samples, they represent the types of current methods that might be used in such an analysis.
Detecting simulated duplications (A) and deletions (B) present in freshwater, but not marine, individuals. We simulated data analogous to our main data set of 10 marine and 11 freshwater stickleback genomes sequenced to a median coverage of 1.7×, except we randomly placed deletions and duplications that ranged from 30 to 1000 bp in the genomes of all freshwater individuals. We recorded the performance of existing methods (at 0.2 false positives) either based on the annotations of individual genomes (cn.MOPS and Genome STRiP) or after we pooled all marine samples into a pseudo-individual and all freshwater samples into a pseudo-individual (rSW-seq and CNVnator). We also tested the transducer's ability to detect heterozygous duplications and deletions. Heterozygous deletions are especially difficult to detect because, with 36-bp reads, regions with SNP divergence may exhibit reduced mapping efficiency, which results in read coverage similar to that of a heterozygous deletion.
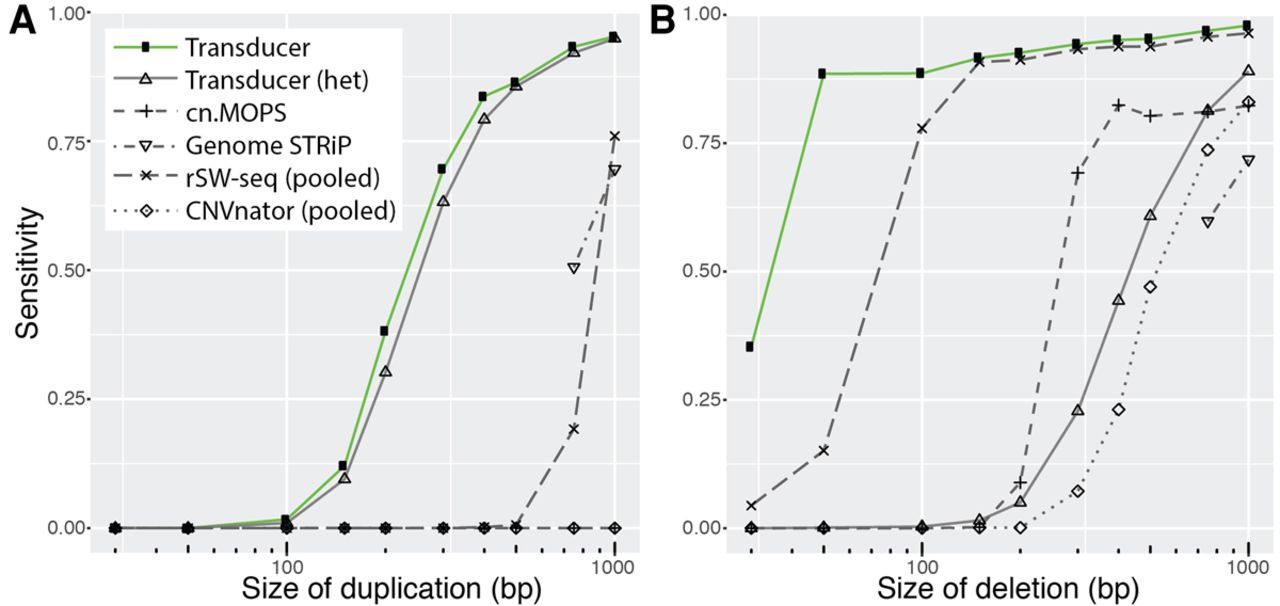
One current method of identifying copy number variants that correlate with group membership is to identify copy number variants in individuals and then perform an additional analysis to test whether the individuals in one group are significantly more likely to have a variant than the other group. We used CNVnator, cn.MOPS, and Genome STRiP to identify CNVs in individuals, followed by Fisher's exact test to identify those regions that correlate with group membership (Supplemental Methods). CNVnator analyzes each sample in isolation and was not able to reliably detect copy number variants <1 kb in our simulated data set. Genome STRiP and cn.MOPS are aware of all samples in the analysis, even when assigning copy number to a specific sample, and this additional information likely improves their performance. In our simulations, both Genome STRiP and cn.MOPS are able to detect CNVs that correlate with group membership, but not with the sensitivity of the transducer presented here (Fig. 3).
An alternative approach with existing tools is to combine the reads from all freshwater individuals into a single pseudo-individual and all reads from marine individuals into a second pseudo-individual. We then used the CNVnator algorithm on each pool and identified regions of the genome where the copy number assigned to one pool differed from the copy number assigned to the other pool (Supplemental Methods). We also used the rSW-seq algorithm on the two pools, because it analyzes exactly two samples at once and identifies regions of differing copy number. On our simulated data sets, the pooling methods performed well, but had reduced sensitivity compared to the transducer presented here (Fig. 3; Supplemental Table S1).
The number of individuals in a group that must deviate from the reference copy number before the model will identify the region as having differential copy number between the two groups is dependent on the sequencing depth of each sample. When simulating deletions in a subset of the freshwater fish in our data set, the model begins to reliably detect a difference between the groups when four of the 10 freshwater fish have deletions compared to zero of the 11 marine individuals.
Validation of deletions and duplications
After testing the transducer on simulated data sets, we first investigated a small subset of the genome in the true data set before identifying copy number variation genome-wide. Although we intend our simulated data sets to be representative of true sequencing libraries, it is likely that some biases and anomalies are not properly represented in the simulated reads. Thus, we used polymerase chain reaction (PCR) followed by gel electrophoresis to test five predicted deletions, and we used quantitative PCR (qPCR) to test four predicted duplications that were identified in this initial analysis of the true data set (Supplemental Tables S2, S3). These validation experiments were performed using DNA samples from a subset of the 21 fish used to make the sequencing libraries and samples from four unrelated individuals. This allowed us to technically validate that the predicted deletions and duplications are in fact present in the sequenced individuals and to biologically validate that consistent differences seen in this sampling of marine and freshwater stickleback are predictive of individuals not yet sequenced. All nine deletions and duplications identified by the model are consistent with the PCR and qPCR results seen in the individuals used to make the sequencing libraries and in the four unrelated individuals (Supplemental Fig. S5). Although these validation experiments are not extensive enough to estimate the rate of false positives at the same resolution as our simulations, they do indicate that the method is able to identify copy number variation that consistently differs between two groups based on low-coverage resequencing data.
Copy number variation correlated with ecotype
Having validated the method with both simulations and PCR-based assays, we applied the transducer genome-wide on the data set of 21 wild-caught marine and freshwater stickleback without masking or excluding any genomic locations. The transducer identified 6664 regions of the genome where the canonical marine and freshwater copy numbers are different (Supplemental Table S4). These regions total 1.7 Mbp (0.4% of the stickleback genome), have an average size of 250 bp, and are distributed across all chromosomes without obvious biases in location (Supplemental Fig. S6). In many cases, the underlying evolutionary events are larger than the regions identified by the model. The three main reasons are (1) in the case of de novo mutations, the model will identify only the shared region that is repeatedly deleted or duplicated, which may be much smaller than some of the individual overlapping events; (2) the model is not aware of splicing, so duplication by processed retrotransposition identifies a separate region for each exon; and (3) deletions and duplications unique to the reference assembly will partition otherwise continuous evolutionary events into multiple regions (Supplemental Fig. S7). By merging regions when they appear in the same protein-coding transcript or if they are within 10 kb of each other, we can address the latter two issues. This enables us to estimate that these 6664 regions are the result of 2643 evolutionary events repeatedly shared by individuals within the same ecotype.
The transducer is not aware of the ancestral state, so it cannot discriminate between insertions in one group versus deletions in the other group. However, we can distinguish those insertion and deletion (indel) events that toggle between the presence and complete absence of a region from those states that signify duplication polymorphisms between one and many copies. In our results, indels are nine times more common than duplications, but this is at least in part a function of our model having more power to detect indels than duplications (Fig. 3). For duplications or indels at least 1kb long, a length that provides sufficient power to detect almost all events in both groups, indels are only twice as prevalent.
These insertions, deletions, and duplications are likely to be of functional significance and under selective pressure to maintain different copy numbers in marine and freshwater populations. This is because alleles following a distribution correlated with an ecological variable represent a departure from what is normally seen for both SNPs and copy number variants, which tend to correlate with geography (Jones et al. 2012; Chain et al. 2014). Based on a Pacific–Atlantic split of our data set, we identified 14,459 regions of copy number variation correlated with ocean basin (approximately 5846 evolutionary events). There are some regions of the genome that give a signal for both ecotypic and geographic variation. These regions tend to represent alleles that are found in freshwater populations in the Pacific, but not Atlantic, region (Supplemental Fig. S8). With so much standing copy number variation in stickleback populations, it is possible that some variants correlated with ecotype could also be due to only sampling 21 populations. When shuffling the group membership of samples 30 times, we identified an average of 2512 regions (maximum 2969 regions) where copy number correlates with a shuffled group assignment. Based on these data, it is likely that some copy numbers correlate with the marine–freshwater ecotype by chance, but that the marine–freshwater set as a whole is not due to the random assignment of standing variation. In an effort to further study the possible functions of the differentiated loci, we analyzed the annotation of these regions, tested sequences experimentally, and examined signatures of selection based on mutational signatures between and within species.
Changes in protein-coding exons
Although it is still difficult to identify the functionally important regions of a genome, protein-coding exons are relatively well annotated and understood. Of the 6664 regions with consistent copy number difference between stickleback ecotypes, 305 overlap protein-coding exons of 211 genes. This includes all 24 genes previously identified as having copy number variants under marine/freshwater parallel selection in a separate study (Hirase et al. 2014), as well as an additional 187 genes.
Relative to the reference genome, 177 of the 211 genes show evidence of indel events in one ecotype (Supplemental Table S5), 72 genes show evidence of duplication (Supplemental Table S6), and 38 genes show evidence of both. These results indicate that >1% of stickleback genes show copy number variation in their protein-coding exons that is repeatedly different between marine and freshwater ecotypes. As would be expected, this group of genes is enriched for having gene expression level differences between marine and freshwater stickleback based on microarray expression surveys (Jones et al. 2012) of multiple adult tissues (P < 10−7). The deletion events tend to affect only one exon of the protein and are enriched for genes involved in the immune system and muscle development (Supplemental Table S7). The duplication events are more likely to include the entire protein and show enrichments for muscle development and immunity and head and limb morphogenesis (Supplemental Table S8). These enrichments for copy number differences are consistent with phenotypic differences that have previously been studied. For example, the stickleback immune system varies with ecotype (Scharsack et al. 2007), marine and freshwater stickleback have different muscle mass and morphology (Dalziel et al. 2012), and marine and freshwater stickleback show consistent morphological differences in their head and limbs (Taylor and McPhail 1986; Caldecutt and Adams 1998; Kimmel et al. 2005). These differences in canonical copy number may thus contribute to many of the phenotypes commonly associated with the divergence of marine and freshwater stickleback.
Changes in gene regulation
More than 95% of the 6664 genomic locations with repeated copy number differences are found outside of coding exons and may affect sequences involved in regulating the expression of nearby genes. Losses of enhancer elements may eliminate tissue-specific expression patterns (Chan et al. 2010), and duplications may make expression patterns more robust to environmental perturbations or increase the level of transcription (Perry et al. 2010). The noncoding regions showing repeated copy number variation are significantly more likely to be located next to a gene involved in the immune system (Supplemental Table S9). This enrichment was also seen for the protein-coding regions and is consistent with known differences between marine and freshwater stickleback populations (Scharsack et al. 2007).
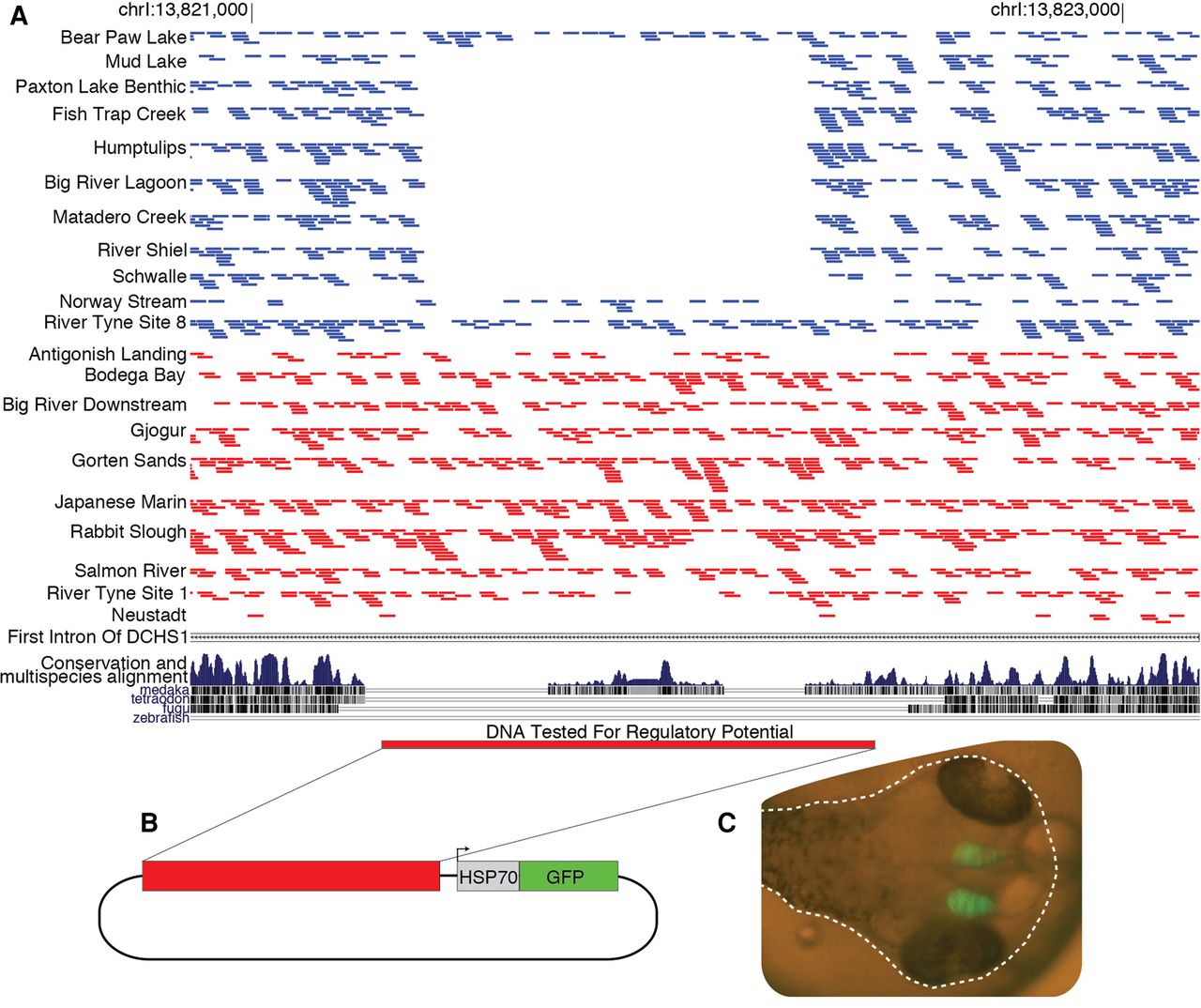
To test whether any of the identified regions function as tissue-specific enhancers, we further studied a region from the DCHS1 gene that is consistently deleted in freshwater fish (Fig. 4; Supplemental Table S10). We cloned the intact noncoding region from a marine stickleback into an expression vector upstream of a minimal promoter attached to an open reading frame for the green fluorescent protein (GFP). Multiple transgenic lines generated with this construct showed GFP expression in the developing telencephalon (Fig. 4). DCHS1 is known to be expressed in the developing telencephalon of zebrafish, mice, and humans (Cappello et al. 2013; B Thisse and C Thisse, unpubl., https://zfin.org/ZDB-PUB-040907-1). Decreasing the expression of Dchs1 in the developing mouse brain leads to additional cell proliferation and differences in neuronal migration (Cappello et al. 2013). Therefore, the loss of the enhancer in freshwater fish could lead to a larger telencephalon. The size and morphology of the telencephalon is highly plastic, but wild-caught freshwater threespine stickleback consistently have larger and morphologically distinct telencephalons compared to those from marine populations (Park and Bell 2010; Park et al. 2012).
Freshwater deletions in DCHS1 remove a conserved sequence that functions as a telencephalon enhancer. (A) The majority of freshwater populations do not have any reads (blue bars) mapping to a region within the first intron of DCHS1, but the marine populations (red bars) appear to universally contain this piece of DNA. The deleted region encompasses an element that shows cross-species conservation with medaka. (B) We cloned three copies of this region upstream of the HSP70 minimal promoter and GFP reporter to test its regulatory potential. (C) We injected this construct into fertilized eggs and multiple transgenic lines show GFP expression in the developing brain at 5 d post-fertilization (dotted white line shows the outline of the embryo within the egg).
Although in vivo assays for enhancer activity provide a wealth of information on the regulatory potential of a DNA segment, the experiments are difficult to scale genome-wide. In contrast, RNA sequencing or microarray analysis makes it possible to test for differential expression of many different genes at particular time points and tissue types. We would expect the set of genes nearest consistent copy number variants to be enriched for genes previously identified as showing marine and freshwater expression differences (Jones et al. 2012). We do detect a highly significant enrichment of marine-freshwater differentially expressed genes, as predicted (P < 10−16). The overall enrichment of 1.2-fold (887 observed with 717 expected) is modest, but many of the regions may be regulating a gene other than the closest transcription start site, and any associated changes in gene expression may affect tissues or time points not represented by the eight adult tissues that were examined.
Another way to identify functional noncoding regions without being limited to a particular tissue or time point is to look for evidence of evolutionary constraint. However, cross-species conservation has its own limitations. Elements must be at least 100 million years old to be found in another sequenced fish (Near et al. 2012), and although constrained elements are functional, many seemingly functional elements show no evidence of constraint (The ENCODE Project Consortium 2007). We created a multispecies alignment and identified 83 Mbp of the genome showing cross-species conservation as indicated by a resistance to substitutions (Supplemental Methods). There are 392 noncoding regions with consistent copy number variation (6% of the set) that overlap regions showing evidence of strong cross-species constraint, suggesting that these regions are likely to have functional consequences if they are deleted or duplicated (Supplemental Tables S11–S13).
Derived alleles in marine stickleback
Stickleback researchers typically view the extant marine populations as representative of the likely ancestral state for recent freshwater populations, since marine phenotypes have stayed relatively static and the marine populations likely coped with environmental changes by shifting their range instead of adapting to a new environment (Bell and Foster 1994). The freshwater populations are viewed as derived, since the populations were likely founded by migratory marine individuals and subsequently underwent large phenotypic transformations in lake and stream habitats created during the glacial retreat (Bell and Foster 1994). In contrast to this view, we detected 86 cases in which the derived deletion alleles are found in marine individuals, whereas freshwater populations maintain sequences conserved to other distantly related teleosts (Supplemental Table S13). These 86 regions are enriched for being near myosin heavy chain genes and ontologies related to the function of this gene family (Supplemental Table S14), perhaps related to differences in swimming performance between marine and freshwater fish (Taylor and McPhail 1986; Law and Blake 1996; Dalziel et al. 2012).
Such deletions may have been positively selected in the ocean after they arose, contributing to their widespread presence throughout marine habitats in the Northern Hemisphere. In this case, there should be reduced heterozygosity flanking the deletions in marine fish but not in freshwater fish that share the ancient allele, and this is what we observe (Supplemental Methods; Supplemental Fig. S9).
The genomic signature of a selective sweep is slowly eroded over time by mutation and recombination, which we may use to estimate when selective sweeps occurred. We simulated sweeps of various ages and found that the overall distribution of heterozygosity flanking the marine deletions resembles that of alleles that originated 100,000 generations ago, although with a more dispersed distribution, consistent with the alleles being created over a range of time rather than all at once (Supplemental Methods; Supplemental Figs. S9, S10). Stickleback typically reproduce once a year and live from one to three years (Bell and Foster 1994), so 100,000 generations corresponds to ∼200,000 yr. Therefore, by the time the last glacial maximum was ending 20,000 yr ago (Clark et al. 2009), these deletion alleles were likely already the predominant alleles in migratory marine fish. How then do we see intact ancestral alleles, without the deletions, in modern post-glacial freshwater populations? One possibility is that ancient intact alleles could spread north from older freshwater populations that inhabit lakes and streams south of the maximum glacial extension, presumably by introgressing and being carried at low frequency in marine populations. To test this hypothesis, we collected hundreds of marine fish from Resurrection Bay in the Gulf of Alaska. We used PCR to screen the marine fish for the presence of intact ancestral alleles at four different loci. We found characteristic intact alleles at all four loci present in the modern marine population, but at low frequencies (0.4%–1.1%) (Supplemental Table S15). This confirms that these ancient alleles are present both in freshwater fish and at a low frequency in marine populations, where they may serve as standing variants that aid in the founding of new freshwater populations.
Discussion
Our study detected copy number variants that show large differences in allele frequency between marine and freshwater ecotypes of a magnitude that is not often observed in other organisms (Hancock et al. 2010; Pritchard et al. 2010). Given ongoing hybridization in contact zones between marine and freshwater fish, strong, but incomplete, selection must occur to maintain characteristic differences between ecotypes against the homogenizing effects of gene flow. Hybridization plays a second important role in the adaptive radiation of sticklebacks: allowing the spread of adaptive alleles between populations. Although previous studies have shown that derived freshwater-adaptive alleles can “hide” at low frequencies in the marine population (Colosimo et al. 2005; Miller et al. 2007; Barrett et al. 2008; Schluter and Conte 2009), our present study suggests this mechanism may also be important for the spread of ancestral alleles between freshwater populations when the derived allele is favored in the marine context. This work in sticklebacks demonstrates that strong selection on either ancestral or derived alleles of standing genetic variation can play an important role facilitating adaptation.
The transducer method presented here should be applicable to many other data sets. For example, we tested the method on a subset of human samples from the 1000 Genomes data set (Sudmant et al. 2015) and were able to detect CNVs not previously annotated by The 1000 Genomes Project Consortium, including two events we successfully confirmed by PCR amplification and sequencing (Supplemental Methods; Supplemental Fig. S11; Supplemental Tables S16, S17). The method presented here may be especially applicable to ancient DNA samples, in which sequence coverage can be sparse and fragment length limits the use of paired-end or mate-pair reads (Green et al. 2010).
There has been great progress in understanding how copy number variation contributes to standing phenotypic variation (Stranger et al. 2007; Yalcin et al. 2011). We leveraged the model system of the stickleback to identify copy number variation that is consistently different between wild vertebrate populations adapting to contrasting environments. We described thousands of regions where copy number is repeatedly different between marine and freshwater stickleback. More than 95% of these regions are likely to affect the regulation of genes since the regions are found outside protein-coding exons. Whether the genes themselves or their regulatory elements are affected by these copy number variants, the functions attributed to the genes align with differences that are consistently reported between marine and freshwater populations: immunity, muscle development, and morphology of the head and limbs. Multiple case studies also implicate adaptive copy number variation in aiding the immunity of humans (Gonzalez et al. 2005; Iskow et al. 2012) and mice (Locke et al. 2015; Pezer et al. 2015), supporting the hypothesis that copy number variation is a general mechanism for adaptation of the vertebrate immune system.
Both copy number variation and base substitutions have contributed to adaptation of humans and other vertebrates (Grossman et al. 2010; McLean et al. 2011; Iskow et al. 2012; Kamberov et al. 2013); however, the relative contributions of the two types of mutations are still unknown. A previous analysis of the same stickleback data set identified 84 regions associated with adaptation to freshwater based on nucleotide substitution patterns (Jones et al. 2012). This is a smaller number than the 6664 regions (approximately 2643 evolutionary events) that we detected for copy number variation. However, a rigorous comparison of the results is difficult, because the two methods have different powers to detect mutations that segregate with ecotype instead of geography. The method presented in this paper can identify a single location of copy number variation that correlates with ecotype instead of geography, whereas the substitution method only has statistical power when blocks of SNPs travel as a unit in marine and freshwater populations. These blocks of continuous marine/freshwater divergence may occur when multiple mutations in the same region contribute to a phenotype or when mutations remain together because of altered recombination patterns, such as in an inversion. It is likely that the results of the substitution screen are a small subset of all substitutions contributing to the repeated colonization of freshwater habitats. The substitution analysis was also constrained to detect only the reuse of standing genetic variation, but the method presented in this paper is also able to detect repeated evolution occurring by different de novo mutations.
Deletions and duplications may produce larger phenotypic effects than single-base changes. This bias toward large effects may increase the probability of copy number mutations being fixed if the corresponding trait is subjected to positive selection within a population, since the probability of fixation is proportional to the magnitude of the mutation's benefit (Haldane 1927).
The ecotypically differentiated copy number variants have a significant overlap with the 84 blocks of marine–freshwater SNP divergence. Fifty-two blocks contain at least one region of repeated copy number variation (P < 10−41). It is possible that some regions of copy number variation within these divergent blocks of DNA are neutral and simply linked with other mutations that are driving selection on the block. However, it is also possible that copy number variants may be contributing to, or be the sole cause of, fitness differences associated with these divergent blocks of DNA.
Although freshwater stickleback populations have been viewed as the ones undergoing large phenotypic changes (Bell and Foster 1994), we identified scores of genomic locations where marine populations have lost conserved ancestral elements that are still present both in freshwater populations and other teleosts, likely due to selective sweeps in the ocean. Viewed as a species complex, stickleback still maintain these ancient conserved noncoding regions, but in an overall ecotypic pattern that reflects selection in the ocean as well as in freshwater environments. Further application of the current method may aid in the detection of many other genomic regions consistently associated with other ecological variables or phenotypic traits, both in stickleback and other species.
Methods
Estimating mappability for each possible read
We estimated the mappability of each k-mer in the reference genome by counting the number of identical k-mers appearing elsewhere in the assembly. If identical reads were the only reason for mismapping, we would expect the probability of a read being mismapped to be
This is likely to be an underestimate since many other factors, such as sequencing errors and differences between the assembly and sequenced individual, can also cause mismapping. Our estimate appears to capture much of the potential for mismapping since it predicts a mismapping rate of 0.08, and simulation shows it to be only slightly higher at 0.09 (Supplemental Methods). We have added a term, ε, which serves to both capture this missing probability of an incorrect mapping and act as a pseudocount so that the mismapping probability in seemingly unique regions of the genome will not be zero.
Solving for ε based on the mismapping rate in simulated data gives a value of 0.01. The performance of the method is not sensitive to small changes in this value (Supplemental Fig. S12). The mismapping equation allows us to estimate both how likely it is that a read generated from a location will mismap elsewhere and how likely a read generated elsewhere will mismap to the given location
Estimating read depth probability
The probability of generating the k-mer being considered (either by reading the bases being analyzed or reading an identical region elsewhere in the genome) is dependent on the copy number at the position being considered, copies (an integer value ranging from 0 to 4 that is defined by the state of the transducer); the number of times the k-mer appears elsewhere in the genome assembly, identicalKmers; and the total number of k-mers (unique in genomic assembly position, but not necessarily sequence) that could be generated from the assembly, t. The value of copies is multiplied by 0.5 to convert the number of copies in a diploid genome to the number of copies that would appear in a genomic assembly. Half copies represent deletions or duplications that are heterozygous in the individual and would therefore appear half or one-and-a-half times in a genome assembly.
We combine Eqs. 3 and 4 to model the probability of a particular read mapping back to the location that would cover the current position. We sum over all k reads that could cover the base to get the probability that a randomly selected read will cover the current base (identicalKmers is now a vector, one position for each of the k reads that could cover the base). This probability is used as the binomial probability for read depth at this specific base in the genome.
Correcting for GC-content
We correct the expected coverage for GC-content because previously published results have shown that reads coming from regions with very low or very high GC-content are underrepresented in sequence data sets (Ross et al. 2013). We apply a correction factor that is specific to each sequencing library because we noticed a significant amount of variability between the libraries used in our study (Supplemental Fig. S13). With reads of length k, there are k+1 bins accounting for all possible GC-contents. We calculate the correction factor for each bin as the fraction of mapped reads with that GC-content divided by the fraction of k-mers in the assembly with the same GC-content. We improve Eq. 5 by adding this GC correction term. gj is the correction factor for the jth read covering the base of interest.
Software availability
Software is available as Supplemental Materials File S1, as well as on GitHub (http://www.github.com/craiglowe).
Data access
New sequencing reads for this study have been submitted to the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA315039. The genomic elements found to be repeatedly different between marine and freshwater stickleback populations are available as Supplemental Table S4.
Acknowledgments
We thank members of the Kingsley Laboratory for useful discussions and comments on the manuscript. Research reported in this publication was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health (K25DE025316), by a Center of Excellence in Genomic Science Grant (National Human Genome Research Institute, HG5P50HG002568 and 3P50HG2568-9S1), and by the National Science Foundation (DEB-0919184). D.M.K. is an Investigator of the Howard Hughes Medical Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.206938.116.
References
- ↵Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984.
- ↵Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE, 2002. Recent segmental duplications in the human genome. Science 297: 1003–1007.
- ↵Barrett RD, Rogers SM, Schluter D. 2008. Natural selection on a major armor gene in threespine stickleback. Science 322: 255–257.
- ↵Bell MA, Foster SA, ed. 1994. The evolutionary biology of the threespine stickleback. Oxford University Press, New York.
- ↵Bell MA, Orti G, Walker JA, Koenings JP. 1993. Evolution of pelvic reduction in threespine stickleback fish: a test of competing hypotheses. Evolution 47: 906–914.
- ↵Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. 2006. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res 8: R38.
- ↵Bradley RK, Holmes I. 2007. Transducers: an emerging probabilistic framework for modeling indels on trees. Bioinformatics 23: 3258–3262.
- ↵Burge C, Karlin S. 1997. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268: 78–94.
- ↵Caldecutt WJ, Adams DC. 1998. Morphometrics of trophic osteology in the threespine stickleback, Gasterosteus aculeatus. Copeia 1998: 827–838.
- ↵Cappello S, Gray MJ, Badouel C, Lange S, Einsiedler M, Srour M, Chitayat D, Hamdan FF, Jenkins ZA, Morgan T, 2013. Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat Genet 45: 1300–1308.
- ↵Chain FJ, Feulner PG, Panchal M, Eizaguirre C, Samonte IE, Kalbe M, Lenz TL, Stoll M, Bornberg-Bauer E, Milinski M, 2014. Extensive copy-number variation of young genes across stickleback populations. PLoS Genet 10: e1004830.
- ↵Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, Brady SD, Southwick AM, Absher DM, Grimwood J, Schmutz J, 2010. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327: 302–305.
- ↵Cheng Z, Ventura M, She X, Khaitovich P, Graves T, Osoegawa K, Church D, DeJong P, Wilson RK, Pääbo S, 2005. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature 437: 88–93.
- ↵Chiang DY, Getz G, Jaffe DB, O'Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES, 2009. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods 6: 99–103.
- ↵Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, Mitrovica JX, Hostetler SW, McCabe AM. 2009. The last glacial maximum. Science 325: 710–714.
- ↵Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM, 2005. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science 307: 1928–1933.
- ↵Dalziel AC, Ou M, Schulte PM. 2012. Mechanisms underlying parallel reductions in aerobic capacity in non-migratory threespine stickleback (Gasterosteus aculeatus) populations. J Exp Biol 215: 746–759.
- ↵Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14: 755–763.
- ↵The ENCODE Project Consortium. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816.
- ↵Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, 2005. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307: 1434–1440.
- ↵Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, 2010. A draft sequence of the Neandertal genome. Science 328: 710–722.
- ↵Grossman SR, Shlyakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, Hostetter E, Angelino E, Garber M, Zuk O, 2010. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 327: 883–886.
- ↵Haldane JB. 1927. A mathematical theory of natural and artificial selection, Part V: selection and mutation. Math Proc Camb Philos Soc 23: 838–844.
- ↵Hancock AM, Alkorta-Aranburu G, Witonsky DB, Di Rienzo A. 2010. Adaptations to new environments in humans: the role of subtle allele frequency shifts. Philos Trans R Soc Lond B Biol Sci 365: 2459–2468.
- ↵Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA. 2015. Large multiallelic copy number variations in humans. Nat Genet 47: 296–303.
- ↵Hirase S, Ozaki H, Iwasaki W. 2014. Parallel selection on gene copy number variations through evolution of three-spined stickleback genomes. BMC Genomics 15: 735.
- ↵Indjeian VB, Kingman GA, Jones FC, Guenther CA, Grimwood J, Schmutz J, Myers RM, Kingsley DM. 2016. Evolving new skeletal traits by cis-regulatory changes in bone morphogenetic proteins. Cell 164: 45–56.
- ↵Iskow RC, Gokcumen O, Lee C. 2012. Exploring the role of copy number variants in human adaptation. Trends Genet 28: 245–257.
- ↵Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484: 55–61.
- ↵Kamberov YG, Wang S, Tan J, Gerbault P, Wark A, Tan L, Yang Y, Li S, Tang K, Chen H, 2013. Modeling recent human evolution in mice by expression of a selected EDAR variant. Cell 152: 691–702.
- ↵Kim TM, Luquette LJ, Xi R, Park PJ. 2010. rSW-seq: algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinformatics 11: 432.
- ↵Kimmel CB, Ullmann B, Walker C, Wilson C, Currey M, Phillips PC, Bell MA, Postlethwait JH, Cresko WA. 2005. Evolution and development of facial bone morphology in threespine sticklebacks. Proc Natl Acad Sci 102: 5791–5796.
- ↵Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U, Hochreiter S. 2012. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res 40: e69.
- ↵Krogh A, Brown M, Mian IS, Sjölander K, Haussler D. 1994. Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol 235: 1501–1531.
- ↵Law T, Blake R. 1996. Comparison of the fast-start performances of closely related, morphologically distinct threespine sticklebacks (Gasterosteus spp.). J Exp Biol 199: 2595–2604.
- ↵Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254.
- ↵Locke ME, Milojevic M, Eitutis ST, Patel N, Wishart AE, Daley M, Hill KA. 2015. Genomic copy number variation in Mus musculus. BMC Genomics 16: 497.
- ↵Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, 1991. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66: 219–232.
- ↵Magi A, Tattini L, Pippucci T, Torricelli F, Benelli M. 2012. Read count approach for DNA copy number variants detection. Bioinformatics 28: 470–478.
- ↵McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, 2011. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471: 216–219.
- ↵Miller CT, Beleza S, Pollen AA, Schluter D, Kittles RA, Shriver MD, Kingsley DM. 2007. cis-Regulatory changes in Kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell 131: 1179–1189.
- ↵Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE. 2006. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res 16: 1182–1190.
- ↵Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65.
- ↵Near TJ, Eytan RI, Dornburg A, Kuhn KL, Moore JA, Davis MP, Wainwright PC, Friedman M, Smith WL. 2012. Resolution of ray-finned fish phylogeny and timing of diversification. Proc Natl Acad Sci 109: 13698–13703.
- ↵Park PJ, Bell MA. 2010. Variation of telencephalon morphology of the threespine stickleback (Gasterosteus aculeatus) in relation to inferred ecology. J Evol Biol 23: 1261–1277.
- ↵Park PJ, Chase I, Bell MA. 2012. Phenotypic plasticity of the threespine stickleback Gasterosteus aculeatus telencephalon in response to experience in captivity. Curr Zool 58: 189–210.
- ↵Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, Werner J, Villanea FA, Mountain JL, Misra R, 2007. Diet and the evolution of human amylase gene copy number variation. Nat Genet 39: 1256–1260.
- ↵Perry MW, Boettiger AN, Bothma JP, Levine M. 2010. Shadow enhancers foster robustness of Drosophila gastrulation. Curr Biol 20: 1562–1567.
- ↵Pezer Z, Harr B, Teschke M, Babiker H, Tautz D. 2015. Divergence patterns of genic copy number variation in natural populations of the house mouse (Mus musculus domesticus) reveal three conserved genes with major population-specific expansions. Genome Res 25: 1114–1124.
- ↵Pritchard JK, Pickrell JK, Coop G. 2010. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol 20: R208–R215.
- ↵Punnett HH, Carpenter GG, Digeorge AM. 1964. Deletion of short arm of chromosome 5. Lancet 2: 588.
- ↵Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. 2013. Characterizing and measuring bias in sequence data. Genome Biol 14: R51.
- ↵Savinainen KJ, Saramaki OR, Linja MJ, Bratt O, Tammela TL, Isola JJ, Visakorpi T. 2002. Expression and gene copy number analysis of ERBB2 oncogene in prostate cancer. Am J Pathol 160: 339–345.
- ↵Scharsack JP, Kalbe M, Harrod C, Rauch G. 2007. Habitat-specific adaptation of immune responses of stickleback (Gasterosteus aculeatus) lake and river ecotypes. Proc Biol Sci 274: 1523–1532.
- ↵Schluter D, Conte GL. 2009. Genetics and ecological speciation. Proc Natl Acad Sci 106: 9955–9962.
- ↵Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, 2005. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77: 78–88.
- ↵Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, 2007. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315: 848–853.
- ↵Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, 2015. Global diversity, population stratification, and selection of human copy-number variation. Science 349: aab3761.
- ↵Taylor EB, McPhail JD. 1986. Prolonged and burst swimming in anadromous and freshwater threespine stickleback, Gasterosteus aculeatus. Can J Zool 64: 416–420.
- ↵Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, 2005. Fine-scale structural variation of the human genome. Nat Genet 37: 727–732.
- ↵Yalcin B, Wong K, Agam A, Goodson M, Keane TM, Gan X, Nellåker C, Goodstadt L, Nicod J, Bhomra A, 2011. Sequence-based characterization of structural variation in the mouse genome. Nature 477: 326–329.