
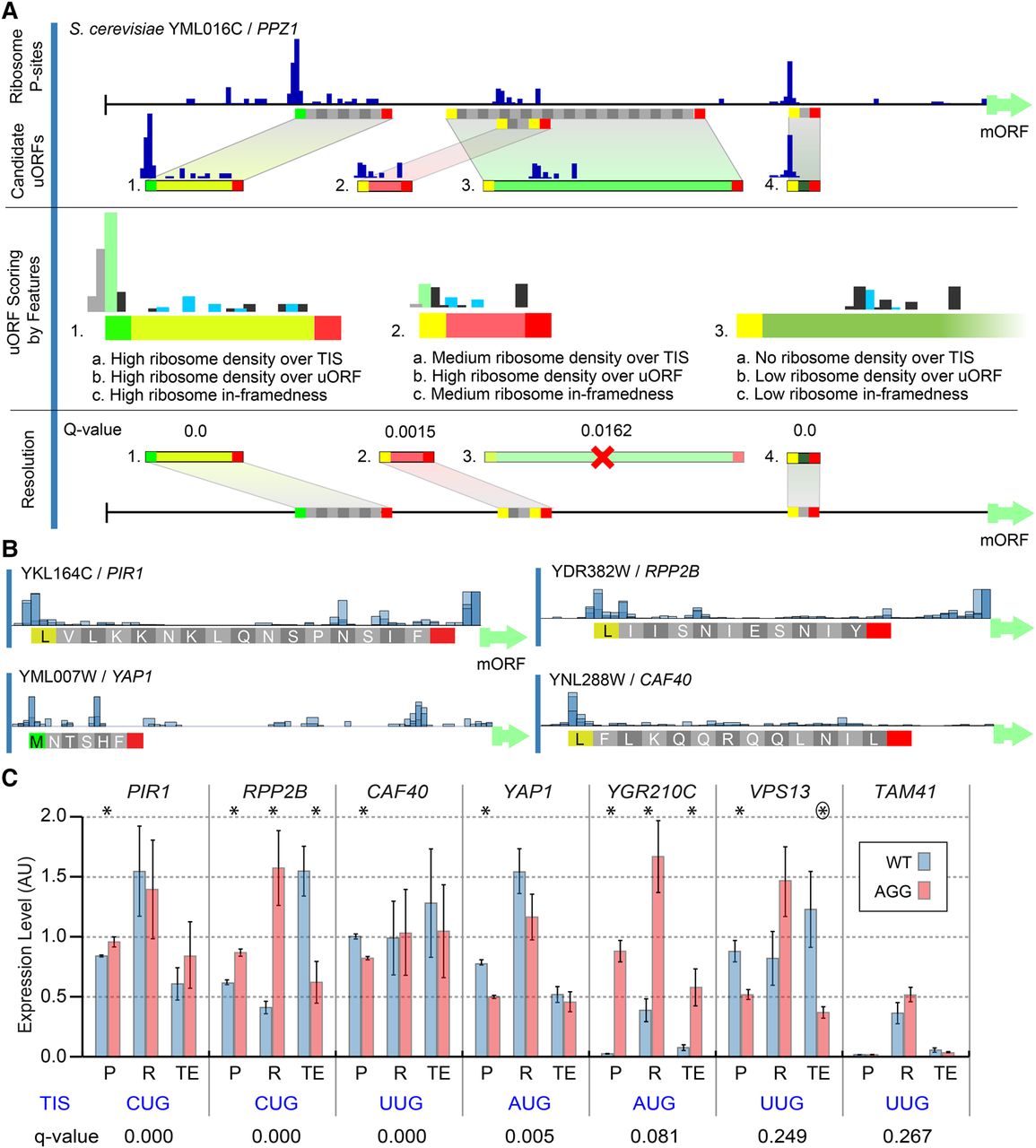
uORF identification using uORF-seqr. (A) uORF-seqr relies on genome annotation, nucleotide sequence, and ribosome profiling and RNA-seq data. PPZ1 (YML016C) is shown as an example. Candidate uORFs (containing potential start and stop codons) are scored for eighteen features (Supplemental Methods) related to Ribo-seq data (P-sites) and uORF position within transcript leaders. A linear regression is used to score each uORF according to the weighted value of these features (Methods), removing candidates that do not score significantly better than null models (Resolution). For overlapping or nested uORFs (e.g., uORFs 2 and 3), the highest scoring candidate uORF is retained. (B) P-site fraction data around four predicted uORFs. Although the Lysine codon is shown, translation is expected to initiate with methionine at NCC codons. (C) Wild-type (AUG or NCC) and AGG start codon mutants were tested for protein (P) and mRNA (R) levels using a dual fluorescence reporter and qPCR, respectively, in triplicate. Bar graphs show the mean and standard error. Estimated translation efficiencies (TE = P/R) were also compared. Significant differences: (*) t-test P < 0.05; (circled asterisk) P = 0.0549.








