
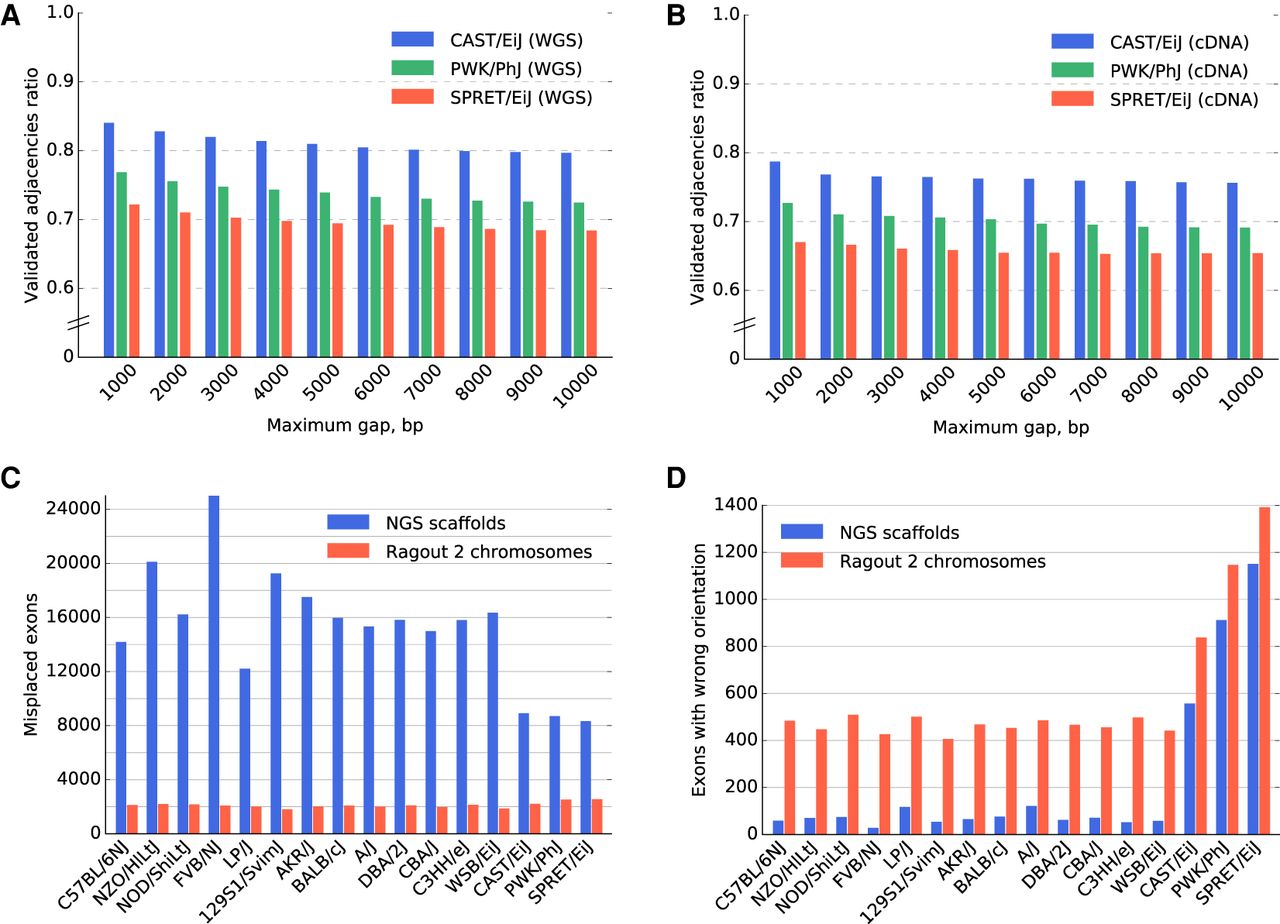
Sixteen mouse laboratory strain assemblies validation. (A,B) The validated adjacency ratio using PacBio reads depending on the maximum gap size of an adjacency for the three most divergent strains. (A) Whole-genome sequence data with approximately 0.5×–1× genome coverage. (B) Whole-exome sequencing with approximately 0.3× genome coverage. The probability of a correct and covered adjacency without a gap not being validated by the reads of length 3000 bp at 1× coverage could be estimated as 15%. (C,D) Ensembl/GENCODE transcript consistency analysis. The genomes are ordered according to the phylogenetic distance from the C57BL6/J reference from the least (C57BL/6NJ) to the most (SPRET/EiJ) divergent. (C) Number of exons found on nonprimary scaffolds/chromosomes. (D) The number of exons on the primary chromosome in a wrong orientation. The total number of transcripts in the database was 78,653. The control analysis of C57BL/6J reference genome yielded 1638 misplaced exons and 517 exons in the wrong orientation (due to ambiguous alignments of short exons). The three most divergent genomes were scaffolded using the Dovetail technology, which explains the increased number of exons with wrong orientation in the NGS scaffolds. Interestingly, the difference between the number of exons with wrong orientation between Ragout 2 chromosomes and NGS scaffolds was lower for the Dovetail-scaffolded genomes.








