
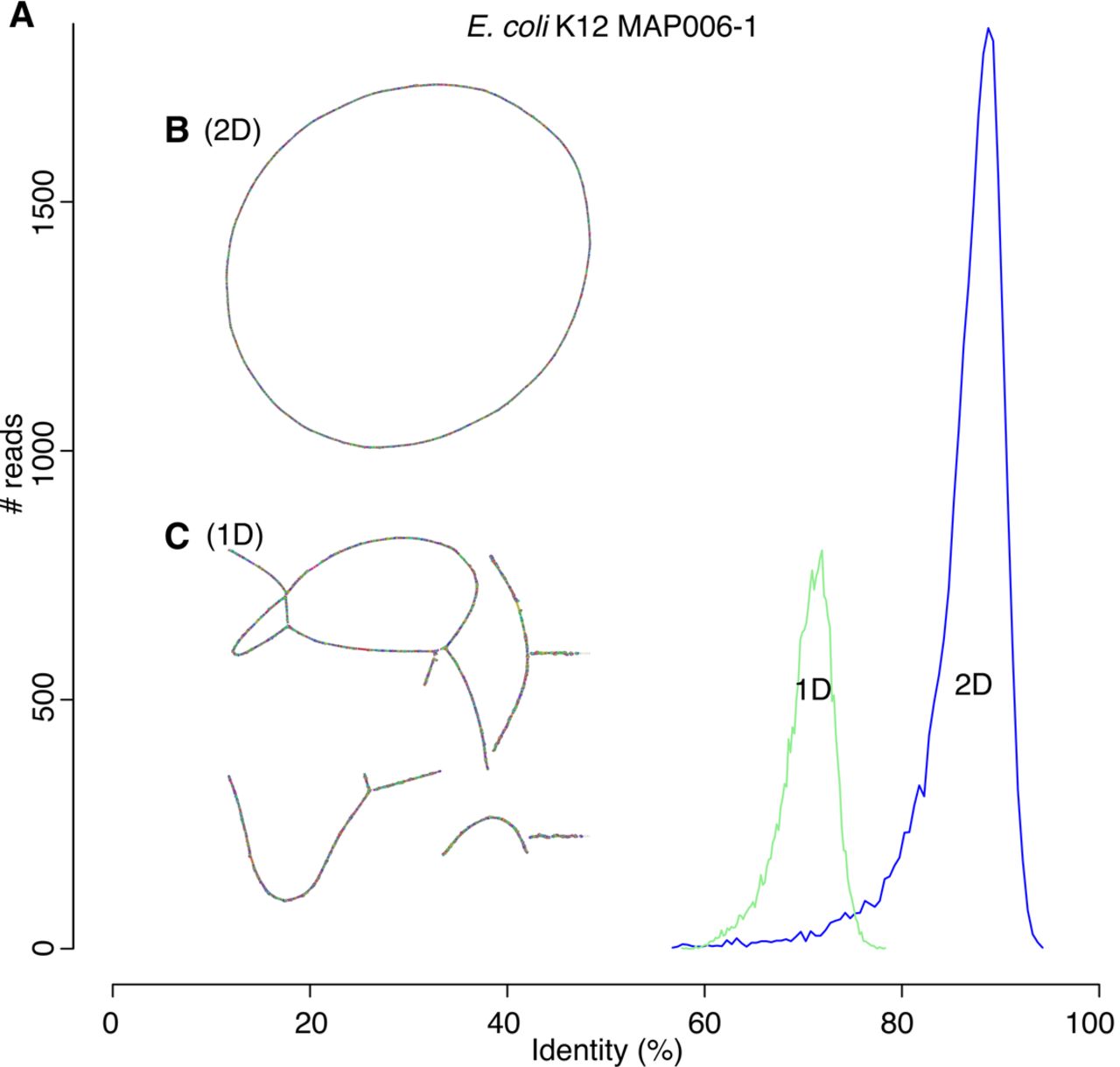
Canu can assemble both 1D and 2D Nanopore Escherhicia coli reads. (A) A comparison of error rates for 1D and 2D read error rates versus the reference. Template 1D and 2D reads from the MAP006-1 E. coli data set were aligned independently to compute an identity for all reads with an alignment >90% of their length (95% of the 2D reads and 86% of the 1D reads had an alignment >90% of their length). The 2D sequences averaged 86% identity, and the 1D reads averaged 70% identity. (B) Bandage plot of the Canu BOG for the 2D data. The genome is in a single circle representing the full chromosome. (C) The corresponding plot for 1D data. While highly continuous, there are multiple components due to missed overlaps and unresolved repeats (due to the higher sequencing error rate).








