
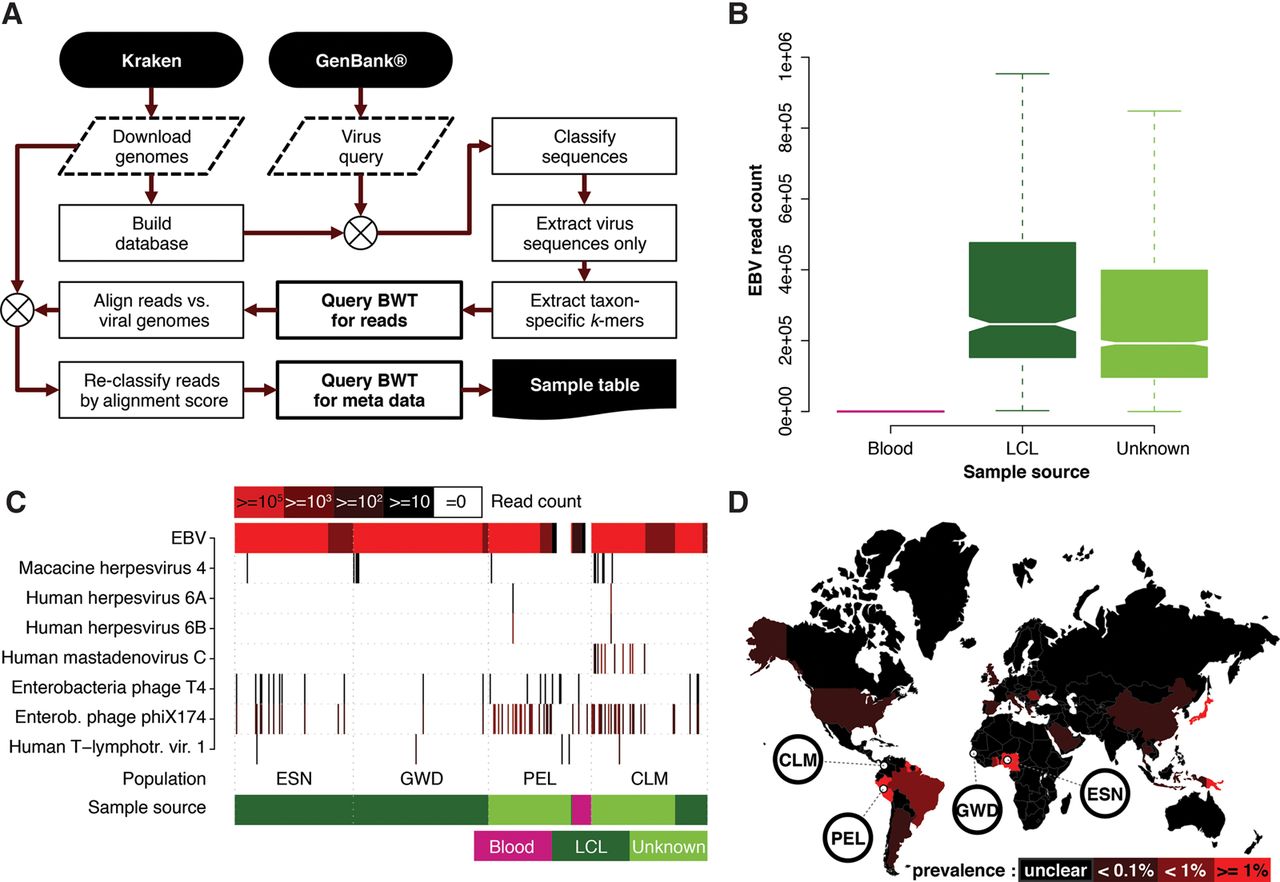
(A) Reference genomes (Human, bacteria, plasmids, and viruses) were downloaded using Kraken's (Wood and Salzberg 2014) built-in routines and a Kraken database generated. GenBank was queried for all virus sequences and the resulting sequence set classified using Kraken to identify taxon-specific 31-mers which were used to query the population BWT for matching reads. Retrieved read sequences were reclassified by alignment to the viral genomes stored in the Kraken database. Finally, sample metadata were retrieved for the final read set. (B) Notched boxplot showing the distribution of human herpesviruses (including EBV) read counts stratified by documented DNA source. Nonoverlapping notches indicate a significant difference of the medians at the 5% level. (C) The populations for which at least one sample contains >10 HTLV-1 reads (black bars) and other virus taxa with >99 reads (red bars) in at least one sample are shown (for all populations, see Supplemental Figs. S3–S8). (D) World map showing HTLV-1 prevalence in different countries, with 1000GP populations that show signal for this virus highlighted.








