
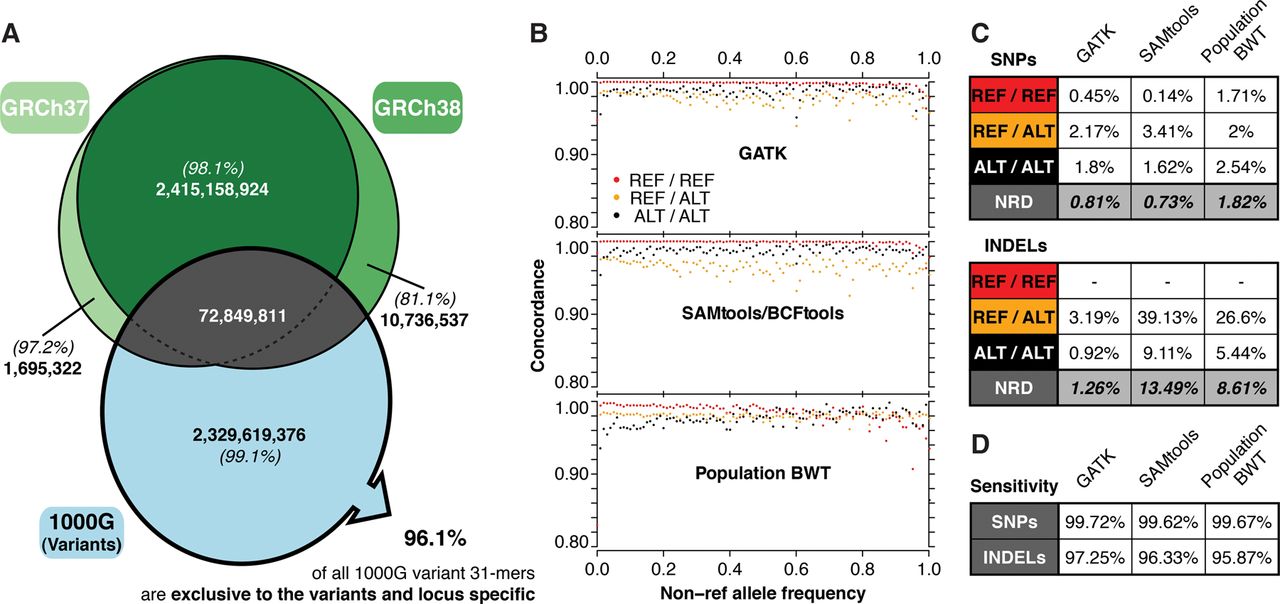
(A) Intersection of the human reference assembly 31-mers and the 1000GP SNP and indel variant 31-mers. The percentages in parentheses give the proportion of these 31-mers that are locus-specific (no other combination of variants in either the same or a different locus in the GRCh37 assembly generates the identical 31-mer). Of all 31-mers generated based on 1000GP variants, 96.1% are locus-specific and exclusive to the variants set, with 91.8% containing a single alternative allele. (B) SNP genotyping of the 1000GP samples at Illumina Omni chip exome-only sites by 31-mer querying of the BWT compared to single sample calling with GATK HaplotypeCaller (v3.5) and SAMtools (v1.1). Dots indicate genotype concordance for variants at different allele frequencies. (C) Genotype discordance rates for SNPs (Omni exome-only: 80,973 sites, all samples) and indels (Genome in a Bottle [Zook et al. 2016] exome in NA12878: 654 sites). (D) Sensitivity of each method expressed as the fraction of total genotypes for which a genotype call was made.








