
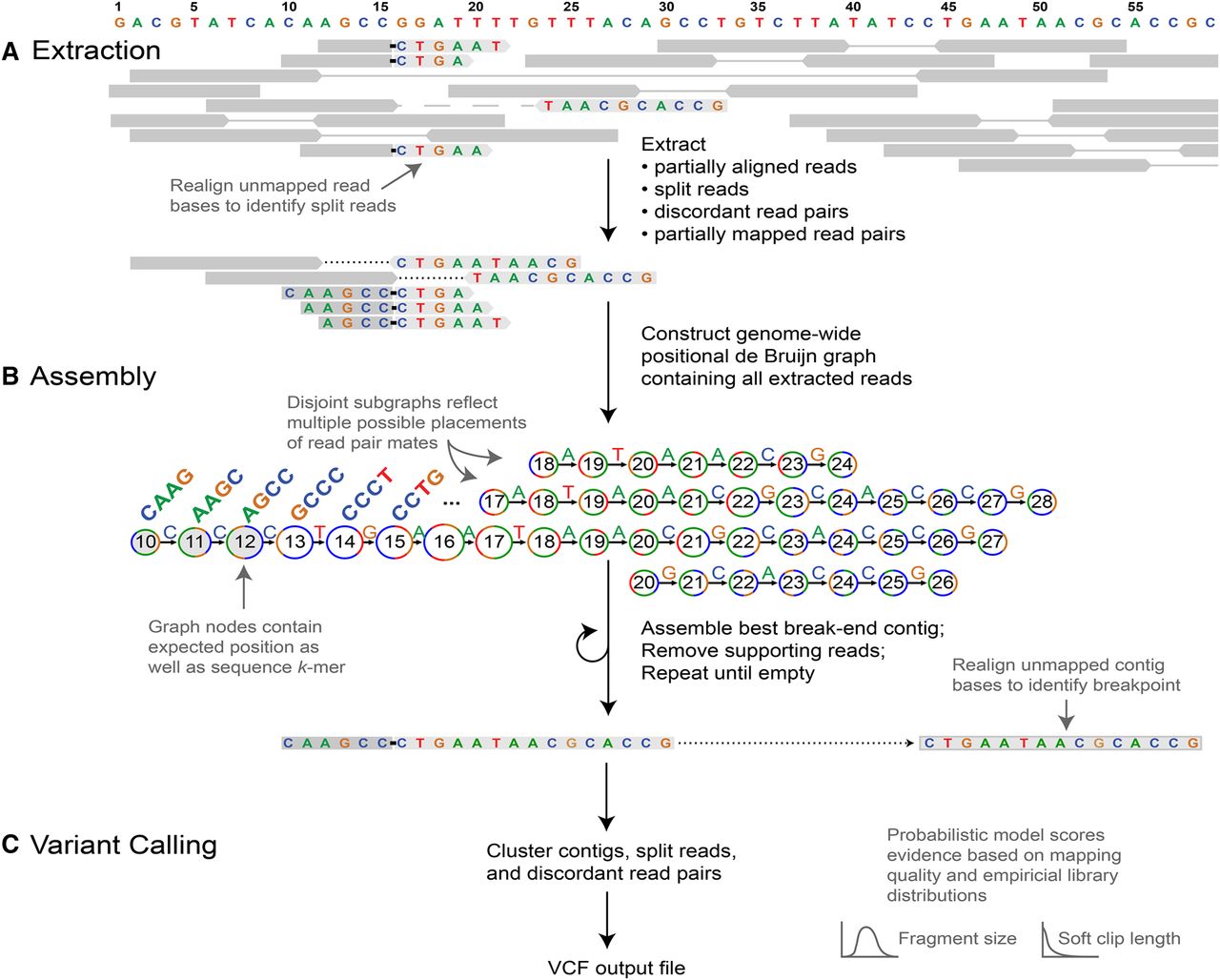
Outline of the GRIDSS pipeline. (A) Soft clipped and indel-containing reads as well as discordant and one-ended anchored read pairs are extracted from input BAM files. Split reads are identified through realignment of soft clipped read bases. (B) Extracted reads are added to a positional de Bruijn graph in all positions consistent with an anchoring alignment. Break-end contigs are identified by iterative identification of the highest weighted unanchored graph path followed by removal of supporting reads. Unanchored contig bases are aligned to the reference genome to identify all breakpoints spanned by the assembly. (C) Variants are called from assembly, split read, and read pair evidence using a probabilistic model to score and prioritize variants.








