
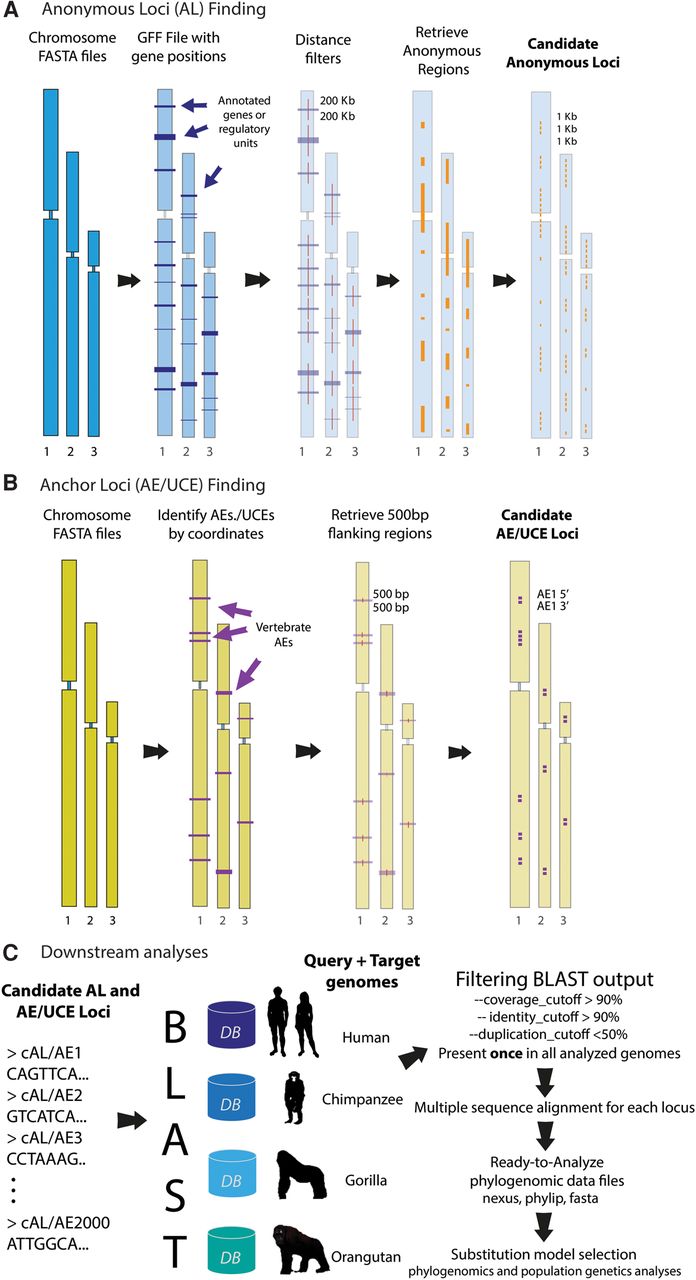
ALFIE software pipeline. (A) Anonymous loci (AL) finding module: User inputs complete genome sequences in a FASTA format and a general feature format (GFF) file for the query genome. Program first applies a user-defined “distance filter,” which removes all known functional elements + flanking sequences of user-specified lengths (purple color blocks). Remaining (presumably neutral) intergenic regions (orange color blocks), called candidate ALs, are retrieved and cut into consecutive segments of user-defined length and saved in FASTA files. (B) Anchor loci (AE/UCE) finding module: User inputs genome sequences in FASTA format. Program finds locations of target AEs/UCEs in a reference human genome with a coordinate file that currently contains 512 vertebrate AEs (included in package). Module retrieves flanking regions with user-defined length (e.g., 500 bp). User also specifies distance (in base pairs) between flanking sequences and their AEs/UCEs. Paired flanking sequences (i.e., candidate AE/UCE loci) are saved in FASTA files. (C) Downstream analyses: AL or AE/UCE candidate loci are used as query sequences in BLAST searches against target genomes. Single-copy loci are retained and subsequently aligned. A user-specified distance filter retains loci that are likely independent from other sampled loci. Each pair of AE/UCE flanking sequences is concatenated to form independent loci. Lastly, ALFIE outputs ready-to-analyze data sets.








