
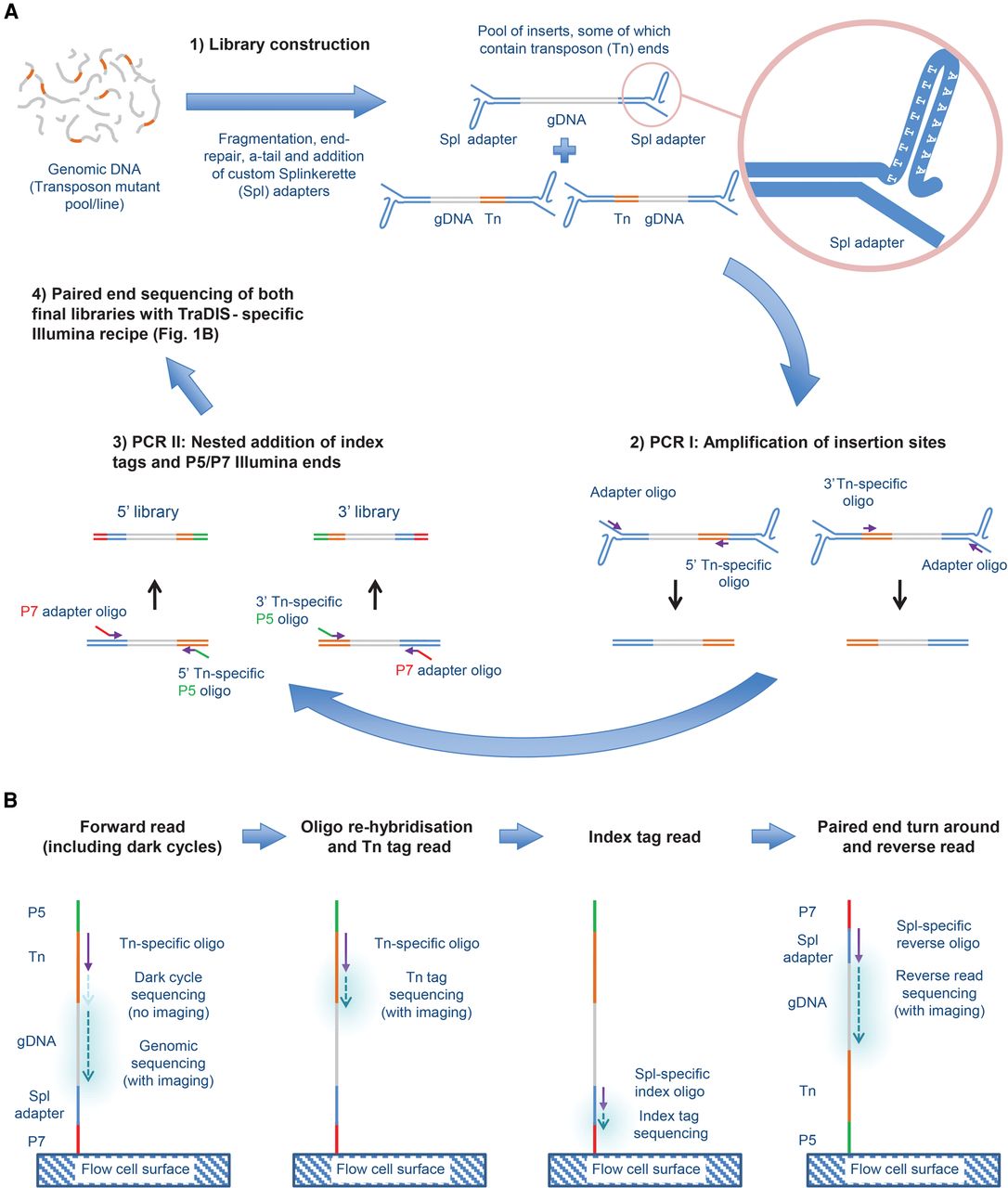
Outline of QIseq process. (A) Library generation. (1) Randomly sheared genomic DNA containing piggyBac transposon (Tn) insertions is ligated to a custom Splinkerette (Spl) adapter (light blue). (2) Separate 5′- and 3′-Tn-end-specific libraries are generated by linear amplification from the transposon (orange) using 5′- or 3′-specific primers, followed by priming from the adapter. (3) A nested PCR is performed with primers specific to the Tn end and adapter end, containing a P5 (green) and P7 (red) sequence, respectively, to allow the product to bind to the Illumina flow cell for sequencing. (B) Sequencing protocol. After binding to the flow cell at the P7 end, a Tn-specific sequencing primer initiates base incorporation. Because all insertion sites will start with the same bases at the end of the Tn, no imaging takes place during the first cycles, with normal imaging initiated only at the genomic DNA sequence. After denaturation, Tn tag sequences are then read using the same sequencing primer, before index tags, which allows sequencing of multiple libraries on one flow cell, and are read using a Splinkerette (Spl)-specific sequencing primer. Finally, a reverse read is generated using a paired-end turnaround, hybridizing the P5 end to the flow cell and sequencing using a Spl-specific reverse oligo.








